
Lesión pulmonar inducida por el ventilador
Complicaciones de la ventilación mecánica: Se resumen los estudios sobre las diversas estrategias de ventilación empleadas para reducir al mínimo la injuria pulmonar, así como las intervenciones farmacológicas.
Autor(es): Dres. Slutsky AS, Ranieri VM.
Enlace: N Engl J Med 2013; 369:2126-36.

Complicaciones de la ventilación mecánica: Se resumen los estudios sobre las diversas estrategias de ventilación empleadas para reducir al mínimo la injuria pulmonar, así como las intervenciones farmacológicas.
Autor(es): Dres. Slutsky AS, Ranieri VM.
Enlace: N Engl J Med 2013; 369:2126-36.

|
El objetivo de la Ventilación mecánica (VM) es proporcionar reposo a los músculos respiratorios al mismo tiempo que brindar el intercambio gaseoso adecuado. A pesar de las indudables ventajas de este tratamiento, muchos pacientes mueren tras la VM, aunque sus gases arteriales quizás se hayan normalizado. Esta mortalidad se atribuye a múltiples factores, entre ellos las complicaciones de la VM, como el barotrauma (i.e., aparición de aire extralveolar), los efectos tóxicos del oxígeno y el compromiso hemodinámico.
Durante la epidemia de polio de 1952 en Copenhagen, se observó que la VM podía causar daño pulmonar estructural. En 1967 se acuñó el término “pulmón de respirador” para describir los infiltrados alveolares difusos y las membranas hialinas halladas en el examen posmortem de pacientes sometidos a VM. Más recientemente, surgió nuevo interés en la lesión que la VM puede producir en pulmones con daño previo y el daño que puede iniciar en pulmones normales.
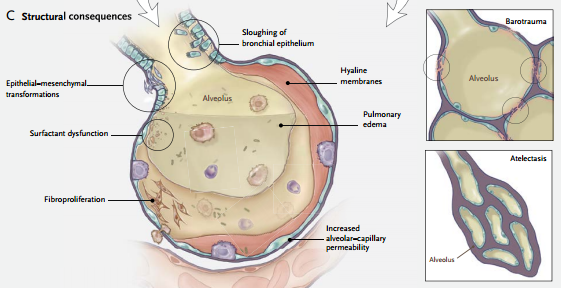
Desde el punto de vista anatomopatológico este daño se caracteriza por infiltrados de células inflamatorias, membranas hialinas, aumento de la permeabilidad vascular y edema pulmonar. El conjunto de las consecuencias pulmonares de la VM se denomina lesión inducida por el respirador.
Recién a principios de este siglo se confirmó la importancia de la lesión pulmonar inducida por el respirador en adultos. Fue cuando un estudio mostró que una estrategia diseñada para reducir al mínimo esta lesión disminuía la mortalidad entre pacientes con síndrome de dificultad respiratoria aguda (SDRA).
Características fisiopatológicas
► Presiones en el pulmón
En cada respiración, la presión necesaria para inflar los pulmones incluye la presión para vencer la resistencia y la inertancia de las vías respiratorias (una medida del gradiente de presión necesario para acelerar el gas) y la presión para vencer las propiedades elásticas del pulmón.
Cuando el flujo aéreo es cero (e.g., al final de la inspiración), la principal fuerza que mantiene la insuflación es la presión transpulmonar (presión alveolar menos presión pleural). Es decir que el volumen pulmonar y la presión transpulmonar están íntimamente unidos.
La sobredistensión pulmonar regional es un factor clave para generar lesión pulmonar inducida por el respirador. Al no haber un método aceptado para medir la sobredistensión regional, se emplea como sustituto la limitación de la presión de insuflación durante la VM a fin de limitar la sobredistensión.
La presión alveolar es relativamente fácil de estimar como la presión de las vías aéreas durante un período de flujo cero; en el paciente sometido a VM que no está efectuando esfuerzos respiratorios espontáneos, la presión de las vías aéreas que se mide durante un período en que se detiene el flujo aéreo al final de la inspiración se denomina presión meseta.
La presión pleural – la otra variable necesaria para calcular la presión transpulmonar- es más complicada. Hay un gradiente gravitacional en la presión pleural que sólo se puede estimar por medición de la presión esofágica. Esta medición es engorrosa y sólo da resultados aproximados. Por eso, la presión meseta es la variable más empleada en la clínica para indicar la sobredistensión pulmonar. Sin embargo, hay matices necesarios al interpretar la presión meseta. Si el paciente no está efectuando esfuerzos respiratorios, la presión meseta representa la presión que está distendiendo los pulmones más la pared torácica.
En el paciente con pared torácica rígida (e.g., el paciente con derrame pleural o ascitis masiva), una gran fracción de la presión proporcionada por el respirador se disipa en insuflar la pared torácica en lugar del pulmón. De modo que la alta presión de las vías aéreas- en este caso, la presión meseta- quizás no sea indicativa de fuerzas de estiramiento pulmonar excesivo (i.e., alta presión transpulmonar).
► Fuerzas físicas
Los siguientes son los principales factores físicos importantes en la producción de lesión inducida por el respirador.
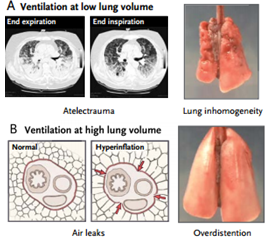
Ventilación a volúmenes pulmonares altos. Se puede producir lesión pulmonar inducida por el respirador debido a ventilación a volúmenes pulmonares altos (absolutos) que lleva a la ruptura alveolar, aparición de aire extralveolar y gran barotrauma (e.g., neumotórax, neumomediastino y enfisema subcutáneo).
El término barotrauma puede ser engañoso, porque la variable esencial que lleva a la aparición de aire extralveolar es la sobredistensión pulmonar regional, no la presión alta de las vías aéreas.
La lesión más sutil que se manifiesta como edema pulmonar se puede producir por la sobredistensión pulmonar. Experimentos efectuados por Webb y Tierney y por Dreyfuss et al mostraron que el volumen (i.e., distensión pulmonar), y no la presión de la vía aérea es el factor más importante que determina la lesión.
Aunque el término de lesión inducida por el respirador es un término aceptado, el nombre puede ser poco apropiado. El factor clave que causa la lesión es la sobredistensión pulmonar, que puede ser causada por otros factores además del respirador.
Ventilación a volúmenes pulmonares bajos. La ventilación a volúmenes pulmonares bajos (absolutos) también puede causar lesión a través de múltiples mecanismos, entre ellos la apertura y el cierre cíclicos de las vías aéreas y las unidades pulmonares, los efectos sobre la función del surfactante y la hipoxia regional. Este tipo de lesión, caracterizada por desprendimiento epitelial, membranas hialinas y edema pulmonar, se denominó “atelectrauma.”
En un estudio clásico, Mead et al, observaron que las fuerzas de distensión en el parénquima pulmonar en los límites entre las zonas aireadas y las zonas atelectásicas podían ser hasta cuatro a cinco veces mayores que las de otras zonas pulmonares.
EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG
|
EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG
|
Fuente: The New England Journal of Medicine
Fuerzas biológicas
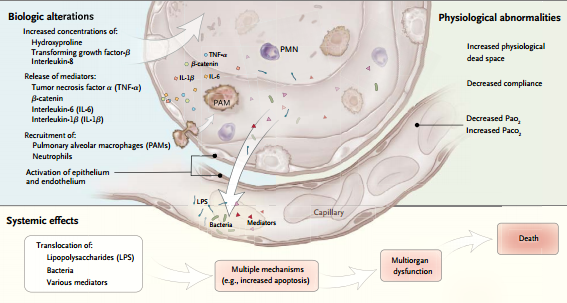
Las fuerzas físicas mencionadas pueden causar la liberación de diversos mediadores intracelulares, ya sea directamente (al lesionar diversas células) o indirectamente (al transducir estas fuerzas en activación de vías de señales en las células epiteliales, endoteliales o inflamatorias). Algunos mediadores pueden lesionar el pulmón directamente; otros pueden preparar el camino para el desarrollo de fibrosis pulmonar.
Otros mediadores pueden actuar como moléculas de migración selectiva directa, reclutando células (e.g., neutrófilos) al pulmón y éstas a su vez pueden liberar más moléculas perjudiciales. Este proceso se denomina biotrauma.
Se puede producir traslocación de mediadores, bacterias o lipopolisacáridos de los espacios aéreos a la circulación sistémica en pulmones que tienen aumento de la permeabilidad alvéolocapilar, que es inherente en el caso del SDRA o que es inducida por volutrauma o microdesgarros epiteliales. Esta traslocación puede causar disfunción multiorgánica y muerte.
EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG
|
Fuente The New England Journal of Medicine
El reconocimiento de la importancia de la lesión pulmonar inducida por el respirador causó un gran cambio en la filosofía de la administración de VM. Mientras que antes el objetivo de la ARM era mantener el intercambio gaseoso a la par que reducir al mínimo el trabajo respiratorio, ahora se estableció otro objetivo: proporcionar el intercambio gaseoso que sostenga la vida mientras se reduce al mínimo la lesión pulmonar inducida por el respirador.
En la práctica, esto significa que programar el respirador a menudo implica disyuntivas difíciles. Por ejemplo, ¿Es mejor emplear un volumen corriente más pequeño y dejar que la presión parcial de anhídrido carbónico arterial (PaCO2) aumente a pesar de los riesgos asociados (e.g., aumento de la hipertensión intracraneal debido a acidosis respiratoria) o emplear volúmenes corrientes mayores para normalizar la PaCO2, pero aumentar el riesgo de lesión pulmonar? En lugar de aumentar el volumen corriente, la filosofía actual se centra más en proteger al pulmón con el empleo de volúmenes corrientes más pequeños.
► Estrategias ventilatorias
A continuación se resumen los estudios sobre las diversas estrategias de ventilación empleadas para reducir al mínimo la lesión pulmonar.
Volúmenes corrientes bajos. Los pacientes con SDRA con frecuencia tienen zonas pulmonares dependientes relativamente no ventiladas (i.e., zonas que desde la perspectiva gravitacional son más bajas que otras zonas y por lo tanto más proclives al colapso) y zonas pulmonares no dependientes ventiladas de manera relativamente normal.
Debido a que el volumen disponible para la ventilación es menor, este trastorno se conoce como “pulmón de lactante”. La consecuencia es que se debería emplear un volumen corriente bajo para prevenir la sobredistensión de las zonas relativamente pequeñas, ventiladas normalmente.
En un estudio fundamental, investigadores de la ARDS Network (Red SDRA) compararon una estrategia de control que empleó un volumen corriente de 12 ml por kilo con una estrategia de volumen corriente bajo que empleó 6 ml por kilo. La estrategia de volumen corriente bajo se asoció con la reducción absoluta de nueve puntos porcentuales de la tasa de muerte (39,8% vs. 31,0%).
PEEP alta y maniobras de reclutamiento. El edema pulmonar y el colapso alveolar de fin de la espiración caracterizan a varias formas de insuficiencia respiratoria. En estas situaciones, la presión positiva espiratoria final (PEEP) baja puede ser insuficiente para estabilizar alveolos y mantenerlos abiertos, aumentando así la probabilidad de lesión pulmonar inducida por el respirador por atelectrauma.
A la inversa, la PEEP más alta puede tener efectos adversos, entre ellos la alteración del retorno venoso y la sobredistensión pulmonar. Un metanálisis reciente de datos de pacientes en estudios aleatorizados investigó estas disyuntivas en pacientes con ARDS y llegó a la conclusión de que una PEEP más alta se asociaba con la reducción absoluta de 5 puntos porcentuales de la tasa de muerte entre los pacientes con peor oxigenación, definida como una relación PaO2:FiO2 de 200 mm Hg o menos.
Dada la importancia de la presión transpulmonar en la lesión pulmonar, el enfoque obvio sería emplear la presión transpulmonar para programar la PEEP, con la presión esofágica como reemplazante de la presión pleural. Sin embargo, la interpretación de la presión esofágica absoluta es difícil debido a artefactos cardíacos, la distribución despareja de la presión pleural (i.e., ningún valor único describe todo el pulmón) y la distorsión y contracción esofágica (especialmente en pacientes en decúbito supino).
No obstante, este enfoque se estudió en pacientes con SDRA. En un estudio piloto, Talmor et al programaron la PEEP para lograr una presión transpulmonar de final de espiración de 0-10 cm de agua, mientras limitaban la presión transpulmonar de final de inspiración a 25 cm de agua. Hallaron mejor oxigenación y una tendencia hacia menor mortalidad a 28 días.
Las maniobras de reclutamiento teóricamente deberían disminuir la lesión pulmonar inducida por el respirador, pero su función en la práctica sigue siendo incierta.
Ventilación de alta frecuencia oscilatoria. La ventilación de alta frecuencia oscilatoria (VAFO) es una técnica en la que volúmenes corrientes muy pequeños (a veces menores que el espacio muerto anatómico) se aplican a altas frecuencias (hasta 15 por segundo). Teóricamente, esta técnica debería ser ideal para reducir al mínimo la lesión pulmonar inducida por el respirador.
En un metanálisis de ocho estudios aleatorizados, controlados, con un total de 419 adultos con SDRA, la mortalidad de los pacientes tratados con VAFO fue significativamente menor que los tratados con la ventilación tradicional (razón de riesgo, 0,77; P = 0,03). Sin embargo, dos estudios multicéntricos recientes con pacientes con SDRA no mostraron mejor evolución con la VAFO, por lo que ésta no puede ser recomendada como tratamiento de primera línea en estos pacientes.
► Estrategias complementarias
Un objetivo de la ARM es contribuir a satisfacer las demandas de intercambio gaseoso del paciente. Un enfoque inespecífico que podría limitar la lesión pulmonar inducida por el respirador es disminuir las demandas metabólicas del paciente y así reducir la ventilación minuto necesaria y disminuir los esfuerzos respiratorios.
Posición en decúbito prono. Alrededor del 70% de los pacientes con SDRA e hipoxemia mejoran la oxigenación cuando se los coloca en decúbito prono. Los posibles mecanismos de este efecto son: aumento del volumen pulmonar de fin de espiración, mejor ajuste ventilación–perfusión, menos efecto de la masa cardíaca sobre los lóbulos inferiores y mejor ventilación regional. Lo más importante es que el decúbito prono minimizaría la lesión pulmonar al aumentar la homogeneidad de la ventilación.
Un metanálisis reciente de siete estudios con un total de 1724 pacientes mostró que el decúbito prono disminuyó la mortalidad absoluta en unos 10 puntos porcentuales en el subgrupo de pacientes con SDRA e hipoxemia intensa (relación PAO2:FIO2, <100 mm Hg).
Los pacientes en decúbito prono sufrieron mayor cantidad de complicaciones prevenibles, entre ellas úlceras por presión, obstrucción del tubo endotraqueal y desplazamiento del mismo. En un estudio reciente con 456 pacientes con SDRA que tenían una relación PAO2:FIO2 <150 mm Hg mientras recibían FIO2 de 0,60 o más, la tasa de muerte a 28 días fue del 32,8% entre aquéllos tratados en decúbito supino y del 16,0% entre los tratados en decúbito prono.
Apoyo extracorpóreo parcial o total. Un enfoque para prevenir la lesión pulmonar inducida por el respirador es evitar la VM y emplear la oxigenación por membrana extracorpórea (ECMO por las siglas del inglés). También se puede combinar la VM con apoyo extracorpóreo parcial. De esta manera, la intensidad de la ventilación necesaria para sostener la vida, disminuye y el anhídrido carbónico se elimina a través de un circuito extracorpóreo. Las ventajas de esta estrategia son la menor tasa de complicaciones, en relación con la ECMO total y la disminución de la lesión pulmonar, al reducir los volúmenes corrientes.
► Intervenciones farmacológicas
Bloqueantes neuromusculares. Debido a la disnea extrema, los pacientes con SDRA a menudo “luchan con el respirador”, lo que puede agravar la lesión inducida por éste. el bloqueante neuromuscular asegura la sincronía paciente-respirador y facilita las limitaciones del volumen corriente y la presión.
En un estudio multicéntrico aleatorizado, controlado con placebo, con 340 pacientes con SDRA y una relación PAO2:FIO2 <150 mm Hg, Papazian et al hallaron que la mortalidad ajustada a 90 días fue más baja entre los que recibieron un bloqueante neuromuscular durante 48 horas que entre los que recibieron placebo.
No se conoce bien el mecanismo preciso del descenso de la mortalidad, pero un estudio anterior mostró la disminución de las citocinas entre pacientes que recibían un bloqueante neuromuscular. En el estudio de Papazian et al, la diferencia de mortalidad entre los grupos se produjo unos 16 días después de iniciado el tratamiento, lo que se podría explicar por la disminución de la insuficiencia multiorgánica debida a biotrauma.
Antinflamatorios y células madre. Estrategias antinflamatorias y el empleo de células madre mesenquimales se investigaron en animales. La principal ventaja de estos tratamientos para prevenir las consecuencias de la lesión pulmonar inducida por el respirador es que el tratamiento se pudo efectuar antes de que el agente desencadenante fuera iniciado (i.e., inmediatamente antes de la VM).
► Aspectos en duda y recomendaciones
Aunque los estudios mencionados ayudan a los médicos a tomar decisiones difíciles, con frecuencia no tienen en cuenta la complejidad de muchas situaciones. En estos casos es aconsejable incorporar los principios fisiológicos a los datos de los estudios. Por ejemplo, en el estudio de la ARDS Network, se empleó la ventilación con un volumen corriente de 6 ml por kilo y se limitó la presión meseta a 30 cm de agua.
Sin embargo, puesto que en algunos pacientes se produce sobredistensión regional, el empleo de un volumen corriente reducido (o PEEP reducida) puede estar justificado. A la inversa, el límite de 30 cm de agua para la presión meseta puede ser demasiado bajo en algunos pacientes (por ejemplo los que tiene una pared torácica muy rígida). En este caso, en un paciente con hipoxemia, pueden ser apropiadas presiones meseta más altas.
Otra posibilidad es medir la presión esofágica para ayudar a programar la estrategia ventilatoria.
Cuando los médicos emplean respiración mecánica que exige la contracción activa de los músculos respiratorios) o ventilación no invasiva, es importante que sepan que se pueden proporcionar volúmenes corrientes grandes a pesar de presiones de las vías aéreas relativamente bajas.
Desde una perspectiva teórica, todos los pacientes en VM se deberían beneficiar con estrategias que reducen al mínimo la lesión pulmonar inducida por el respirador. No obstante, es importante tener en cuenta los efectos de cualquier estrategia para disminuir la lesión pulmonar sobre otros fenómenos fisiológicos o clínicos (e.g., el efecto de una estrategia que protege el pulmón sobre la hemodinamia). Interesa especialmente saber si los pacientes con pulmones relativamente normales deben ser sometidos a ventilación con volúmenes corrientes bajos.
Los pulmones normales toleran volúmenes corrientes relativamente grandes administrados a presiones relativamente bajas siempre y cuando el estrés y las presiones aplicadas estén por debajo del umbral de lesión. Los umbrales específicos son inciertos, pero un metanálisis reciente mostró que la ventilación con volúmenes corrientes más pequeños en pacientes sin SDRA puede estar asociada con mejores resultados. Aunque estos datos sugieren que las estrategias de VM que protegen el pulmón se deben adoptar ampliamente, la estrategia ideal aún no se ha determinado.
1. Lassen HC. A preliminary report on the 1952 epidemic of poliomyelitis in Copenhagen with special reference to the treatment of acute respiratory insufficiency. Lancet 1953; 1:37-41.
2. Nash G, Blennerhassett JB, Pontoppidan H. Pulmonary lesions associated with oxygen therapy and artificial ventilation. N Engl J Med 1967; 276:368-74.
3. Macklin MT, Macklin CC. Malignant interstitial emphysema of the lungs and mediastinum as an important occult complication in many respiratory diseases and other conditions: an interpretation of the clinical literature in the light of laboratory experiment. Medicine 1944; 23:281-358.
4. Avignon PD, Hedenstrom G, Hedman C. Pulmonary complications in respirator patients. Acta Med Scand Suppl 1956; 316: 86-90.
5. Respirator lung syndrome. Minn Med 1967; 50:1693-705.
6. Fothergill J. Observations of a case published in the last volume of the medical essays of recovering a man dead in appearance, by distending the lungs with air. Philosophical Transactions 1744; 43: 275-81.
7. The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med 2000; 342:1301-8.
8. Rahn H, Otis AB, Chadwick LE, Fenn WO. The pressure-volume diagram of the thorax and lung. Am J Physiol 1946;146: 161-78.
9. Zin WA, Milic-Emili J. Esophageal pressure measurement. In: Hamid Q, Shannon J, Martin J, eds. Physiologic basis of respiratory disease. Hamilton, ON, Canada:BC Decker, 2005: 639-47.
10. Bouhuys A. Physiology and musical instruments. Nature 1969; 221:1199-204.
11. Ware LB, Matthay, MA. The acute respiratory distress syndrome. N Engl J Med 2000;342: 1334-49.
12. Slutsky AS, Hudson LD. PEEP or no PEEP — lung recruitment may be the solution. N Engl J Med 2006; 354:1839-41.
13. Maunder RJ, Pierson DJ, Hudson LD. Subcutaneous and mediastinal emphysema: pathophysiology, diagnosis, and management. Arch Intern Med 1984; 144: 1447-53.
14. Webb HH, Tierney DF. Experimental pulmonary edema due to intermittent positive pressure ventilation with high inflation pressures: protection by positive end expiratory pressure. Am Rev Respir Dis 1974; 110:556-65.
15. Dreyfuss D, Soler P, Basset G, Saumon G. High inflation pressure pulmonary edema: respective effects of high airway pressure, high tidal volume, and positive end-expiratory pressure. Am Rev Respir Dis 1988;137:1159-64.
16. Mascheroni D, Kolobow T, Fumagalli R, Moretti MP, Chen V, Buckhold D. Acute respiratory failure following pharmacologically induced hyperventilation: an experimental animal study. Intensive Care Med 1988;15:8-14.
17. Slutsky AS. Lung injury caused by mechanical ventilation. Chest 1999; 116:Suppl:9S-15S.
18. Bilek AM, Dee KC, Gaver DP III. Mechanisms of surface-tension-induced epithelial cell damage in a model of pulmonary airway reopening. J Appl Physiol 2003; 94:770-83.
19. Albert RK. The role of ventilation induced surfactant dysfunction and atelectasis in causing acute respiratory distress syndrome. Am J Respir Crit Care Med 2012; 185:702-8.
16. Mascheroni D, Kolobow T, Fumagalli R, Moretti MP, Chen V, Buckhold D. Acute respiratory failure following pharmacologically induced hyperventilation: an experimental animal study. Intensive Care Med 1988;15:8-14.
17. Slutsky AS. Lung injury caused by mechanical ventilation. Chest 1999; 116:Suppl:9S-15S.
18. Bilek AM, Dee KC, Gaver DP III. Mechanisms of surface-tension-induced epithelial cell damage in a model of pulmonary airway reopening. J Appl Physiol 2003; 94:770-83.
19. Albert RK. The role of ventilation induced surfactant dysfunction and atelectasis in causing acute respiratory distress syndrome. Am J Respir Crit Care Med 2012; 185:702-8.
20. Mead J, Takishima T, Leith D. Stress distribution in lungs: a model of pulmonary elasticity. J Appl Physiol 1970; 28:596-608.
21. Tremblay L, Valenza F, Ribeiro SP, Li J, Slutsky AS. Injurious ventilatory strategies increase cytokines and c-fos m-RNA expression in an isolated rat lung model. J Clin Invest 1997; 99:944-52.
22. Cabrera-Benítez NE, Parotto M, Post M, et al. Mechanical stress induces lung fibrosis by epithelial-mesenchymal transition. Crit Care Med 2012; 40:510-7.
23. Tremblay LN, Slutsky AS. Ventilator induced injury: from barotrauma to biotrauma. Proc Assoc Am Physicians 1998; 110:482-8.
24. Ranieri VM, Suter PM, Tortorella C, et al. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: a randomized controlled trial. JAMA 1999; 282: 54-61.
25. Nahum A, Hoyt J, Schmitz L, Moody J, Shapiro R, Marini JJ. Effect of mechanical ventilation strategy on dissemination of intratracheally instilled Escherichia coli in dogs. Crit Care Med 1997; 25:1733-43.
26. Murphy DB, Cregg N, Tremblay L, et al. Adverse ventilatory strategy causes pulmonary-to-systemic translocation of endotoxin. Am J Respir Crit Care Med 2000;162: 27-33. [Erratum, Am J Respir Crit Care Med 2002; 166:1517.]
27. Slutsky AS, Tremblay LN. Multiple system organ failure: is mechanical ventilation a contributing factor? Am J Respir Crit Care Med 1998; 157:1721-5.
28. Gattinoni L, Pesenti A. The concept of “baby lung.” Intensive Care Med 2005; 31:776-84.
29. Hickling KG, Henderson SJ, Jackson R. Low mortality associated with low volume pressure limited ventilation with permissive hypercapnia in severe adult respiratory distress syndrome. Intensive Care Med 1990; 16:372-7.
30. Amato MB, Barbas CS, Medeiros DM, et al. Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med 1998;
338:347-54.
31. Briel M, Meade M, Mercat A, et al. Higher vs lower positive end-expiratory pressure in patients with acute lung injury and acute respiratory distress syndrome: systematic review and meta-analysis. JAMA 2010; 303:865-73.
32. Talmor D, Sarge T, Malhotra A, et al. Mechanical ventilation guided by esophageal pressure in acute lung injury. N Engl J Med 2008; 359:2095-104.
33. Lachmann B. Open up the lung and keep the lung open. Intensive Care Med 1992; 18:319-21.
34. Mascia L, Pasero D, Slutsky AS, et al. Effect of a lung protective strategy for organ donors on eligibility and availability of lungs for transplantation: a randomized controlled trial. JAMA 2010; 304:2620-7.
35. Brower RG, Morris A, MacIntyre N, et al. Effects of recruitment maneuvers in patients with acute lung injury and acute respiratory distress syndrome ventilated with high positive end-expiratory pressure. Crit Care Med 2003; 31:2592-7. [Erratum, Crit Care Med 2004; 32:907.]
36. Ferguson ND, Slutsky AS. High-frequency ventilation is the optimal physiological approach to ventilate ARDS patients. J Appl Physiol 2008; 104:1230-1.
37. Sud S, Sud M, Friedrich JO, et al. High frequency oscillation in patients with acute lung injury and acute respiratory distress syndrome (ARDS): systematic review and meta-analysis. BMJ 2010; 340:c2327.
38. Ferguson ND, Cook DJ, Guyatt GH, et al. High-frequency oscillation in early acute respiratory distress syndrome. N Engl J Med 2013; 368:795-805.
39. Young D, Lamb SE, Shah S, et al. High-frequency oscillation for acute respiratory distress syndrome. N Engl J Med 2013; 368:806-13.
40. Malhotra A, Drazen JM. High-frequency oscillatory ventilation on shaky ground. N Engl J Med 2013; 368:863-5.
41. Piedalue F, Albert RK. Prone positioning in acute respiratory distress syndrome. Respir Care Clin N Am 2003; 9:495-509.
42. Broccard A, Shapiro RS, Schmitz LL, Adams AB, Nahum A, Marini JJ. Prone positioning attenuates and redistributes ventilator-induced lung injury in dogs. Crit Care Med 2000; 28:295-303.
43. Valenza F, Guglielmi M, Maffioletti M, et al. Prone position delays the progression of ventilator-induced lung injury in rats: does lung strain distribution play a role? Crit Care Med 2005; 33:361-7.
44. Sud S, Friedrich JO, Taccone P, et al. Prone ventilation reduces mortality in patients with acute respiratory failure and severe hypoxemia: systematic review and meta-analysis. Intensive Care Med 2010; 36:585-99.
45. Guérin C, Reignier J, Richard J-C, et al. Prone positioning in severe acute respiratory distress syndrome. N Engl J Med 2013; 368:2159-68.
46. Brodie D, Bacchetta M. Extracorporeal membrane oxygenation for ARDS in adults. N Engl J Med 2011; 365:1905-14.
47. Gattinoni L, Kolobow T, Agostoni A, et al. Clinical application of low frequency positive pressure ventilation with extracorporeal CO2 removal (LFPPV-ECCO2R) in treatment of adult respiratory distress syndrome (ARDS). Int J Artif Organs 1979;2:282-3.
48. Terragni PP, Del Sorbo L, Mascia L, et al. Tidal volume lower than 6 ml/kg enhances lung protection: role of extracorporeal carbon dioxide removal. Anesthesiology 2009; 111:826-35.
49. Bein T, Weber-Carstens S, Goldmann A, et al. Lower tidal volume strategy (approximately 3 ml/kg) combined with extracorporeal CO2 removal versus “conventional” protective ventilation (6 ml/kg) in severe ARDS: the prospective randomized Xtravent-study. Intensive Care Med 2013; 39:847-56.
50. Slutsky AS. Neuromuscular blocking agents in ARDS. N Engl J Med 2010; 363:1176-80.
51. Papazian L, Forel JM, Gacouin A, et al. Neuromuscular blockers in early acute respiratory distress syndrome. N Engl J Med 2010; 363:1107-16.
52. Forel JM, Roch A, Marin V, et al. Neuromuscular blocking agents decrease inflammatory response in patients presenting with acute respiratory distress syndrome. Crit Care Med 2006; 34:2749-57.
53. Uhlig S, Uhlig U. Pharmacological interventions in ventilator-induced lung injury. Trends Pharmacol Sci 2004; 25:592-600.
54. Curley GF, Hayes M, Ansari B, et al. Mesenchymal stem cells enhance recovery and repair following ventilator-induced lung injury in the rat. Thorax 2012; 67:496-501.
55. Grasso S, Stripoli T, De Michele M, et al. ARDS net ventilatory protocol and alveolar hyperinflation: role of positive end-expiratory pressure. Am J Respir Crit Care Med 2007; 176:761-7.
56. Futier E, Constantin J-M, Paugam-Burtz C, et al. A trial of intraoperative low-tidal-volume ventilation in abdominal surgery. N Engl J Med 2013; 369:428-37.
57. Serpa Neto A, Cardoso SO, Manetta JA, et al. Association between use of lungprotective ventilation with lower tidal volumes and clinical outcomes among patients without acute respiratory distress syndrome: a meta-analysis. JAMA 2012;308: 1651-9.

No hay comentarios:
Publicar un comentario