
Síndromes coronarios agudos
Mecanismos de formación y ruptura de la placa ateromatosa: Una revisión exhaustiva y rigurosa del conocimiento acerca de la fisiopatología de los síndromes coronarios agudos. Aterogénesis y la intimidad del "accidente de placa coronaria".
Autor(es): Jacob Fog Bentzon, Fumiyuki Otsuka, Renu Virmani, Erling Falk
Enlace: © American Heart Association, Inc.,April 7, 2014.
Enlace: © American Heart Association, Inc.,April 7, 2014.

|
Bases y limitaciones del conocimiento actual
Lo que hoy sabemos sobre los mecanismos de la enfermedad aterosclerótica se deriva principalmente de la patología descriptiva de autopsias humanas y de la patología experimental en modelos animales con hipercolesterolemia grave. En esta revisión introductoria, no vamos a discutir cómo se obtuvo de cada pieza de la información y los puntos fuertes y las limitaciones de los estudios subyacentes, pero vamos a señalar algunas advertencias generales de nuestra base de conocimientos.
Los modelos animales son la piedra angular para la comprensión de una enfermedad tan compleja como la aterosclerosis, pero los modelos sólo están disponibles para el desarrollo de la lesión asintomática y no para los procesos que conducen a la trombosis.
Otra cuestión posiblemente relacionada es que los modelos actuales, de las que el ratón transgénico es el más ampliamente utilizado, esencialmente están modelando la hipercolesterolemia familiar homocigótica con un modo acelerado de la progresión de la enfermedad durante meses, mientras que en la vida real, la enfermedad progresa durante muchas décadas.
La patología de la aterosclerosis en el extremadamente pequeño subgrupo de pacientes con hipercolesterolemia familiar homocigótica parece diferir del proceso de la enfermedad multifactorial común en varios aspectos importantes, incluyendo una menor frecuencia de trombosis como causa de muerte.14 Por lo tanto, puede que no sea sorprendente que las características morfológicas y el destino de las lesiones sean muy diferentes en los modelos animales y en los seres humanos.
Estas limitaciones de la investigación se reflejan en lo que sabemos sobre la enfermedad.
Existe un mayor conocimiento mecanicista en profundidad de cómo el colesterol LDL provoca la formación de la lesión aterosclerótica, pero mucho se sabe menos acerca de cómo otros factores de riesgo importantes, como la hipertensión, el tabaquismo o la diabetes mellitus, están involucrados, y por qué algunas placas, pero no otras, finalmente causan complicaciones trombóticas devastadoras.
Un aumento de la concentración en sangre de las lipoproteínas que contienen apolipoproteína B, de las cuales la lipoproteína de baja densidad (LDL) por lo general es la forma más frecuente, puede ser una causa suficiente de la aterosclerosis, tales como en la hipercolesterolemia familiar y otras hiperlipidemias genéticas (enfermedad monofónica).
Más a menudo, sin embargo, la enfermedad se desarrolla en los niveles más bajos de LDL en combinación con otros factores de riesgo que facilitan la aterosclerosis (enfermedad multifactorial).6,7 Estos incluyen el tabaquismo, la hipertensión, la diabetes mellitus, el sexo masculino, y una susceptibilidad genética compleja a la enfermedad (antecedentes familiares).
El hecho de que las enfermedades del corazón se puedan prevenir de varias maneras diferentes, por ejemplo con las estatinas y con el uso de drogas antihipertensivas, dejar de fumar u otras modificaciones de estilo de vida, es un reflejo de su base multifactorial.
Las personas con muy bajo LDL generalmente no desarrollan aterosclerosis clínicamente relevante, independientemente de la presencia de otros factores de riesgo.8 Además, los estudios de aleatorización mendeliana que investigan el efecto potencial de la reducción del LDL durante toda la vida han demostrado los efectos protectores de los niveles de LDL bajos heredados, lo que subraya la importancia fundamental del LDL como un factor causal también para la forma común, multifactorial de la enfermedad.9 Juntos, los factores de riesgo modificables conocidos explican más del 90% de la aparición de IAM en las poblaciones de todo el mundo.6
Aunque los mecanismos fisiopatológicos centrales se supone que son los mismos con independencia del conjunto de factores etiológicos en un paciente en particular, la presencia de factores de riesgo individuales añade matices a la presentación de la enfermedad.
Por ejemplo, el consumo de cigarrillos aumenta el riesgo de IAM más que el de la angina de pecho estable,10 la hipertensión es un poderoso factor de riesgo excepcional para el accidente cerebrovascular,11 y el tabaquismo y la diabetes mellitus representan la mayor parte del riesgo de desarrollar enfermedad vascular periférica.12 El nivel de LDL parece ser menos importante para el accidente cerebrovascular y para la enfermedad vascular periférica que para la enfermedad coronaria.12,13 Los niveles plasmáticos de LDL parece ser menos importantes para el riesgo de ACV y enfermedad vascular periférica que para la enfermedad coronaria.12,13
La enfermedad coronaria y otras manifestaciones de la aterosclerosis no se encontraban entre las causas más comunes de muerte hasta el comienzo del siglo XX, pero a partir de entonces se registró un dramático aumento en los países industrializados, incluyendo Europa occidental y los Estados Unidos, con un pico alrededor de 1.960 a 1.980.1
Aumentos comparables en la incidencia de las enfermedades del corazón se han producido más tarde en muchas otras partes del mundo, principalmente debido al crecimiento demográfico y al aumento de la expectativa de vida media.2 La aterosclerosis es hoy en día a escala mundial la principal causa de muerte y discapacidad con la mayor carga de la enfermedad en el mundo en desarrollo.2
En los países ricos, la mortalidad ajustada por edad por enfermedad coronaria ha disminuido considerablemente en las últimas décadas. Sin embargo, esta disminución se explica en parte por la mejora de la supervivencia después de un infarto de miocardio (IAM) ,y a medida que más individuos se encuentran en riesgo de desarrollar enfermedades del corazón debido a un envejecimiento de la población, la prevalencia y la carga económica de estas enfermedades es probable que se incremente.3-5 Por lo tanto, la tarea es más importante que nunca para los investigadores y para los clínicos cardiovasculares es trabajar hacia su prevención, sobre todo con estrategias que se pueden aplicar en todo el mundo.
▶ Mecanismos de formación de la placa ateroesclerótica
Los mecanismos de la enfermedad provocados por las LDL y por los otros factores causales son múltiples como se discute más abajo, lo que implica la retención de la lipoproteína, el reclutamiento de células inflamatorias, la formación de células espumosas, la apoptosis y la necrosis, la proliferación de las células del músculo liso y la síntesis de matriz, la calcificación, la angiogénesis, la remodelación arterial , la ruptura de la capa fibrosa, la trombosis, y más.
Hay una compleja interacción entre estos procesos y una variable de importancia de cada proceso en el desarrollo de las placas individuales que conducen a tasas de progresión impredecibles, morfología de la placa heterogénea y resultados clínicos variables. La mayoría de las placas permanecen asintomáticas (enfermedad subclínica), algunos se convierten en obstructivas (angina estable) y otras provocan trombosis aguda y pueden dar lugar a un síndrome coronario agudo (SCA).
La patogénesis de las lesiones ateroscleróticas se ha deducido a partir del análisis microscópico de las arterias en diferentes grupos de edad. Esta área de investigación ha sido ampliamente revisado por Stary et al15-17 en una serie de artículos que siguen siendo uno de los recursos más ricos para las descripciones microscópicas de la aterosclerosis. Este trabajo también dio lugar a una clasificación histológica propuesta de las lesiones (Clasificación Asociación Americana del Corazón, los tipos I-VIII).18
Una clasificación alternativa y más simple, que hace hincapié en el vínculo entre la morfología de la lesión y la enfermedad clínica, fue introducida más tarde por Virmani et al19 y es utilizada en el presente artículo. Los principales tipos de lesiones reconocidas en esta clasificación se muestran en la Figura 1. Un solo paciente con enfermedad avanzada podrá albergar típicamente muchos tipos diferentes de lesiones en diferentes sitios del árbol arterial.
 Figura 1. Tipos de lesiones de aterosclerosis y una secuencia propuesta de su desarrollo. Una, adaptable engrosamiento de la íntima que se caracteriza por la acumulación de células de músculo liso dentro de la íntima. B, de la íntima xantoma correspondiente a la acumulación de macrófagos de células de espuma dentro de la íntima. Engrosamiento de la íntima patológicos en C denota la acumulación de piscinas de lípidos extracelulares en la ausencia aparente de la necrosis. D, fibroateroma que indica la presencia de un núcleo necrótico. El núcleo necrótico y el tejido circundante pueden eventualmente estar calcificadas, que forma la placa fibrocalcificada muestra en E. Debido a que algunos de los tipos de lesiones avanzadas (fibroateromas y placas fibrocalcificadas) Evoluciona de forma simultánea en la vida, sus interrelaciones son difíciles de resolver en los estudios de autopsia. Movat pentacromo mancha. EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG |
La aterosclerosis es una enfermedad multifocal que ataca regiones reproducibles del árbol arterial. Los sitios con baja tensión de cizallamiento oscilatoria endotelial (shear stress), los situados cerca de los puntos de ramificación y a lo largo de las curvaturas interiores, son los más susceptibles.20
La aorta abdominal, las arterias coronarias, las arterias iliofemorales y las bifurcaciones carotídeas son normalmente los más afectados. Antes del desarrollo de la aterosclerosis, estos sitios de predilección se caracterizan por cambios en el volumen endotelial y en la expresión génica, presencia de células dendríticas subendoteliales,20, 21 y, en seres humanos, por la presencia de engrosamiento de la íntima por adaptación (Figura 1A).15, 22
Los engrosamientos adaptativos de la íntima se desarrollan de forma espontánea después del nacimiento y pueden llegar a ser tan gruesos como la media subyacente y proporcionar un terreno para el desarrollo inicial de la lesión.15, 22 La tasa de progresión sigue siendo más altas aquí que en otros sitios arteriales. Sin embargo, con el tiempo, la enfermedad se propaga a la íntima adyacente,23 y en pacientes de edad avanzada que mueren de IAM, la mayor parte de las arterias coronarias epicárdicas se ven afectados por placas.24
Los experimentos en animales indican una relación causal entre la baja tensión de cizalla de la pared y el inicio de la enfermedad,25 pero el mecanismo de mediación no se entiende completamente. Cuando la placa se desarrolla, la pared arterial se remodela, los patrones de flujo locales cambian. Esta interacción dinámica entre el flujo y la pared del vaso puede influir en la progresión de la enfermedad y en última instancia, en el destino de las lesiones.
▶ Mecanismos de ruptura de la placa
▶ Mecanismos de erosión
Un patrón no uniforme especial de tipo denso de tipo I (mayores) y laxo III (menores) de colágeno, juzgada para indicar ruptura de la placa curada, puede ser identificado en muchas placas coronarias, especialmente en aquellos que causan la estenosis crónica de alto grado (Figura 6).104 a menudo varios sitios de ruptura curadas están presentes, y el número de rupturas curadas se correlaciona con la gravedad de stenosis.105 Otras placas pueden tener una apariencia de múltiples capas consistente con una historia de incorporación y organización de trombo en plaques.19 erosionados Aunque estos resultados son observacional, indican que la placa se rompe en silencio y erosiones son importantes para el crecimiento de la placa y la formación de estenosis crónica (Figura 3). Por otra parte, similar a una cicatriz contracción del tejido fibroso de cicatrización se ha propuesto como una causa de la remodelación constrictiva menudo visto con plaques.84 severamente estenótica,114 Un papel central de la curación de la placa en la formación de estenosis puede explicar por qué la estenosis coronaria crónica a menudo se desarrolla en un lugar fásica de manera lineal, formando en los sitios que sólo se redujeron de manera insignificante en un angiografía.104, 115
▶ Inflamación mediada por las lipoproteínas
Las moléculas de LDL causan la aterosclerosis mediante su acumulación en la íntima arterial donde pueden ser modificadas por oxidación y agregación26 Las LDL modificadas y los restos lipídicos oxidados derivados de ellas, a su vez actúan como estimuladores crónicas de la respuesta inmune innata y adaptativa. Inducen a las células endoteliales y las células musculares lisas a expresar moléculas de adhesión (por ejemplo, moléculas de adhesión celular vascular y moléculas de adhesión intercelular), quimioatrayentes (por ejemplo, proteína quimiotáctica de los monocitos-1), y factores de crecimiento (por ejemplo, factor estimulante de colonias de macrófagos y factor estimulante de colonias de macrófagos de granulocitos) que interactúan con los receptores de los monocitos y estimular la migración y diferenciación en macrófagos y células dendríticas.27, 28
La unión de la LDL a los proteoglicanos de la íntima es un paso importante para el inicio de la enfermedad,29 que potencialmente puede explicar la propensión a la aterosclerosis de engrosamientos adaptativos de la íntima. A medida que la enfermedad evoluciona, sin embargo, el endotelio se vuelve más permeable, y la expresión de moléculas de unión de lipoproteínas-en la placa puede promover aún más la capacidad de retener LDL.30 Esto sugiere que se necesitan niveles más altos de LDL para inducir la enfermedad que para mantener la progresión una vez que las lesiones ya se han formado, una idea coherente con la fuerte relación entre los niveles de LDL en la edad adulta joven y el riesgo de desarrollar enfermedades del corazón más tarde.30-32
Los macrófagos reclutados expresan varios fenotipos diferentes de polarización y ejercen múltiples efectos en el desarrollo de la lesión.33 Algunos macrófagos alcanzan un fenotipo proinflamatorio M1, posiblemente a través de la unión de la LDL modificada a los receptores de reconocimiento de patrones (por ejemplo, los receptores Toll-like), y segregan citoquinas proinflamatorias (por ejemplo, interleucina-1β y el factor de necrosis tumoral-α), enzimas, y especies reactivas de oxígeno que promueven aún más la retención y la modificación de LDL (por ejemplo, mieloperoxidasa), así como muchos otros mediadores que han demostrado desempeñar un papel en la aterosclerosis (por ejemplo, los activadores del plasminógeno, catepsinas, y metaloproteinasas de matriz).33 Otros tienen un fenotipo M2 y pueden secretar factores (por ejemplo, la transformación del factor de crecimiento β y lípidos pro-resolución) que favorecen la resolución de la inflamación.33-36
El sistema inmune adaptativo reconoce a la molécula de LDL modificada y a otros autoantígenos relacionados con el proceso aterosclerótico, y las células inmunes participan en el desarrollo de lesiones.27 Los linfocitos T helper 1 aparecen en la íntima coincidentemente con las células espumosas y secretan citoquinas proinflamatorias (interferón-γ y factor de necrosis tumoral-α) que acentúan la inflamación vascular en modelos de ratón,37 pero otros tipos de células inmunes, tales como las células T reguladoras y células B, posiblemente la mejoren.38
Ambos, macrófagos y células dendríticas sirven como depósitos de lípidos de las lipoproteínas infiltradas y se convierten en células espumosas por mecanismos que aún no se entienden bien in vivo.39 Pueden implicar la absorción mediada por el receptor de la LDL oxidada, la hidrólisis de colesterol libre a partir de complejos de LDL agregados en compartimientos extracelulares líticos, la captación directa de la LDL nativa o de las lipoproteínas remanentes, o de hecho una combinación de estos y otra vías propuestas.40-42 Notablemente, las células musculares lisas también toman lípidos en la aterosclerosis humana y se acumulan gotitas de ésteres de colesterol, posiblemente por mecanismos similares.43
Las células espumosas, fácilmente reconocibles por microscopía de luz, son indicadores luminosos de la inflamación impulsada por lipoproteínas en la pared vascular. En un principio se acumulan dentro de la capa de proteoglicanos de la íntima, y cuando ya se han formado varias capas, son visibles a simple vista como xantomas de color amarillo o estrías grasas (fig. 1B).
Los xantomas son inofensivos y completamente reversibles si los estímulos que provocaron su formación se disipan. Están presentes ya en algunas aortas fetales y en recién nacidos en los primeros 6 meses de vida, lo que probablemente refleja los factores de riesgo de la madre,44 pero su número disminuye en años posteriores. En la adolescencia, reaparecen en las regiones propensas a la aterosclerosis de las arterias coronarias y de la aorta en la mayoría de las personas.45
Muchos xantomas no progresan más, pero algunos, especialmente los que ocurren en los sitios de predilección, se convierten en lesiones ateroscleróticas progresivas. La característica definitoria es la acumulación de material acelular rico en lípidos en la íntima. Las acumulaciones de lípidos más pequeños son vistas inicialmente por debajo de las capas de células espumosas y sin interrupción grave de la estructura normal de la íntima.16, 19, 46 Este tipo de lesión se denomina engrosamiento intimal patológico (Figura 1C) y se observa comúnmente a los 20 a 30 años de edad en la arterias coronarias.16, 47
En algunas lesiones, las acumulaciones de lípidos aisladas se convierten en núcleos necróticos confluentes (núcleo rico en lípidos) a través de la invasión de macrófagos. Este proceso altera irreversiblemente la estructura normal de la íntima y deja tras de sí una matriz gelatinosa de lípidos y restos celulares.46
El núcleo necrótico puede ser caracterizado tarde o temprano por la siguiente formación que muestra una cierta presencia de ácido hialurónico y versican con infiltración de macrófagos.48 Cuando un núcleo necrótico está presente la lesión es un fibroateroma.
La apoptosis y la necrosis secundaria de las células espumosas y de las células musculares se cree que son una causa importante para el desarrollo del núcleo necrótico.49, 50 Muchos factores que son capaces de inducir la apoptosis in vitro están presentes en las placas,51, 52 y es razonable suponer que varios de éstos cooperan para causar apoptosis en los macrófagos y en las células musculares lisas in vivo.49
Los macrófagos apoptóticos y las células musculares lisas se hacen detectables de manera coincidente con la aparición de acumulaciones de lípidos en las lesiones humanas.53 La muerte celular por apoptosis, tanto como por otras formas de muerte celular, se puede ver en el margen del núcleo necrótico.54 La presencia de restos apoptóticos libres en las lesiones, es decir no asociados con células fagocíticas, indica que la eliminación deteriorada de los restos apoptóticos (eferocitosis) contribuye al crecimiento del núcleo necrótico.36, 55
Esta teoría del desarrollo del núcleo necrótico se apoya en experimentos en ratones utilizando la ingeniería genética para inducir o proteger contra la apoptosis en los macrófagos y en las células musculares lisas. En las primeras etapas de la aterosclerosis, la inhibición selectiva de la apoptosis de los macrófagos aumenta el número de células espumosas, lo que indica que la apoptosis, aunque indetectable en esta etapa debido a un eferocitosis eficiente, mantiene el balance del reclutamiento y proliferación de los macrófagos en curso.56
Sin embargo, cuando es inducida experimentalmente y después de que se ha comenzado a formar los focos necróticos, la apoptosis de los macrófagos y de las células musculares lisas aumenta necrótico núcleo en la formación de la placa y la inflamación,56, 57 posiblemente porque los fagocitos vecinos ya no eliminan de manera eficiente a los remanentes apoptóticos.36 En lugar de ello, se dejan que la necrosis secundaria y la carga rica en lípidos se deposite en el tejido donde se incita aún más a la inflamación.36, 56, 57
La composición química del núcleo necrótico indica que otras fuentes de lípidos pueden ser también contribuyentes importantes, incluyendo la acumulación directa de ésteres de colesterol a partir de LDL58 y membranas de eritrocitos ricos en colesterol libre derivados de hemorragias intraplaza.48 También es posible que la hidrólisis extracelular de macrófagos catalizada por agregados de LDL pueda contribuir al alto contenido de colesterol libre en el núcleo necrótico.59
Por qué se produce necrosis en algunas, pero no otras lesiones, no se conoce y, al parecer, los factores causales están al menos parcialmente disociados de los que causan los xantomas. Por ejemplo, los hombres y las mujeres desarrollan una cantidad similar de xantomas coronarios tempranos en la vida, pero los hombres tienen muchas lesiones ateroscleróticas más progresivas a la edad de 30 a 60 años.
▶ Angiogénesis y hemorragia intraplaca
Los neovasos procedentes esencialmente de los vasa vasorum de la adventicia se convierten en la base de las lesiones ateroscleróticas progresivas y una vía de entrada alternativa para los monocitos y las células inmunes con una importancia cuantitativa desconocida.61 Los neovasos de la placa carecen de células de soporte y son frágiles y con fugas, dando lugar a extravasación local de proteínas de plasma y eritrocitos.62 Tales hemorragias intra-placa son comunes en los fibroateromas y pueden ampliar el núcleo necrótico y promover inflamación.48 Otra fuente común de hemorragia de la placa es la extravasación de sangre a través de la ruptura de una capa fibrosa.64
Nuevas datos indican que la respuesta inflamatoria a la hemorragia intraplaca se acentúa en los pacientes con deterioro de la función de haptoglobina causado por el genotipo común HP2-2 incluyendo en particular a los pacientes con diabetes mellitus,66 y esto puede contribuir al aumento del riesgo de enfermedad coronaria en individuos con elevada hemoglobina glicosilada.67
El tejido conectivo de las lesiones es inicialmente el de la íntima arterial normal o un engrosamiento adaptativo de la íntima, pero poco a poco este tejido fibrocelular perdido se sustituye por tejido fibroso rico en colágeno que a menudo crece hasta convertirse en el componente cuantitativamente dominante de los tejidos de las placas.68 El tejido que se encuentra entre el núcleo necrótico y la superficie luminal de la placa (capa fibrosa) es fibroso y con un alto contenido de colágeno de tipo I.
El colágeno, elastina, y proteoglicanos de la matriz fibrosa es producido principalmente por las células musculares lisas. Las células musculares lisas de la placa se caracterizan por un retículo de Golgi y abundante complejo endoplásmico rugoso y sólo escasos miofilamentos.69 Este fenotipo se ha denominado sintético en contraste con el fenotipo contráctil de las células musculares lisas mediales.
Las calcificaciones son comunes en las lesiones ateroscleróticas progresivas y aumentan con la edad. Las células apoptóticas, matriz extracelular y material del núcleo necrótico pueden actuar como nido para gránulos microscópicos de calcio que posteriormente se ampliarán hasta formar grumos más grandes de los depósitos de calcio.77, 78 El núcleo necrótico puede calcificarse por completo con el tiempo y esas calcificaciones pueden constituir la mayor parte de su volumen.17
Durante la aterogénesis, el segmento de vaso local tiende a remodelarse de manera tal que el área de la luz por lo general no se ve comprometida hasta que las placas son grandes (remodelación expansiva)81 A partir de entonces las estenosis pueden ocurrir a través del crecimiento continuo de la placa o de la contracción del segmento local (remodelación constrictiva) o de una combinación de ambos procesos.81, 82 La remodelación expansiva es más frecuente en los fibroateromas, y el grado de ampliación se correlaciona positivamente con la inflamación de la placa, la atrofia medial, y el tamaño del núcleo necrótico.83, 84 Los segmentos con remodelación constrictiva contienen a menudo lesiones ricas en tejido fibroso.84
El modo y el alcance de la remodelación es al menos tan importante como tamaño de la placa en la determinación de la gravedad de la estenosis. Por lo tanto, las imágenes de la luz de una arteria por angiografía no son útiles para el diagnóstico de la presencia de una placa aterosclerótica, y viceversa, el examen microscópico de las placas no puede estimar el grado de estenosis luminalin vivo.
▶ Mecanismos de la erosión, ruptura y trombosis de la placa
La aterosclerosis solo puede obstruir el flujo sanguíneo coronario y causar angina de pecho estable, pero esto rara vez es fatal en ausencia de cicatrices del miocardio que pueden provocar una arritmia o muerte cardíaca súbita. Los síndromes coronarios agudos están casi siempre causados por un trombo luminal o una hemorragia repentina de la placa aterosclerótica, con o sin vasoespasmo concomitante.
En el infarto de miocardio con elevación del segmento ST la trombosis es en su mayoría oclusiva y sostenida. Mientras que en la angina inestable y en el infarto sin supradesnivel del ST el trombo suele ser incompleto y dinámico, o incluso inexistente.
También en víctimas de muerte súbita coronaria, se encuentra con frecuencia la trombosis aguda u organizada; el resto muere con enfermedad coronaria severa en ausencia de trombosis con o sin cicatrices miocárdicas.19, 85
Causas más raras de síndromes coronarios agudos son los émbolos, la disección de la arteria, la vasculitis, el abuso de la cocaína, los puentes de las arterias coronarias y el trauma.
La ruptura de la placa es la causa más frecuente de trombosis.85, 86 En la ruptura de la placa, un defecto estructural -una brecha en la capa fibrosa- expone el núcleo altamente trombogénico a la sangre. El material de la placa desalojada a veces se encuentra dentro del trombo, lo que indica que la ruptura y la trombosis coincidían y por lo tanto apoya su relación causal.
 Figura 2. Una trombosis causada por ruptura de la placa. La placa responsable se muestra en A es un fibroateroma que consiste en tejido fibroso (F), áreas dominadas por las acumulaciones de lípidos extracelulares (PT), y los núcleos necróticos plenamente desarrollados (NC). B, Grande ampliación del recuadro naranja en A. La capa fibrosa fina e inflamada que cubre el gran núcleo necrótico se ha roto y material de la base, incluyendo cristales de colesterol (*), ha sido impulsado hacia la luz, donde se puede encontrar en la base de el trombo. Tinción de elastina-tricrómico (azul colágeno). EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG |
En casos raros, las calcificaciones nodulares (nódulo calcificado) sobresalen hacia la luz a través de una capa fibrosa rota, y esto se ha sugerido como un mecanismo desencadenante de trombosis.19 Cuando no es posible identificar una rotura de la placa a pesar de una búsqueda exhaustiva microscópica, se utiliza el término “erosión” de la placa término. Este término se eligió debido a que el endotelio está típicamente ausente debajo del trombo pero, si este es el mecanismo de precipitación sigue siendo desconocido.19 Tanto el engrosamiento de la íntima patológica como los fibroateromas se pueden complicar por erosión de la placa.
En una reciente recopilación de datos procedentes de los estudios de autopsias en todo el mundo, la mayoría de los trombos coronarios fatales se asociaron con la ruptura de placa, independientemente de la presentación clínica (IAM, el 79%, muerte súbita coronaria, 65%), edad (> 60 años, el 77% ; <60 años, 64%; desconocido, el 73%), sexo (hombres, 76%; mujeres, 55%), y los continentes (Europa, 72%; Estados Unidos, el 68%; Asia, 81%).88
La ruptura de la placa también es el sustrato más común para los trombos que causan infarto de miocardio en aquellos que sobreviven.89 Las diferencias de sexo parecen dignas de mención y, curiosamente, la ruptura de la placa se ha encontrado que es particularmente frecuente en las mujeres premenopáusicas, que sin embargo constituyen un muy pequeño grupo de víctimas de ataques al corazón.90
Algunos estudios han informado de que la diabetes mellitus, el tabaquismo y el nivel de la hiperlipidemia están asociados con el mecanismo de la trombosis en los síndromes coronarios agudos pero, salvo el sexo y la menopausia, no hay relaciones consistentes que hayan sido demonstrated.88 Fumar parece promover la trombosis coronaria, independientemente del tipo de placa.91 Un reciente estudio clínico utilizando tomografía de coherencia óptica intravascular indicó que la aparición ruptura de la placa puede ser mayor en el IAM con elevación del segmento ST que en los infartos sin elevación del segmento ST.92, 107

Figura 3. Ruptura de la placa y la curación. La rotura de un fibroateroma capitalización delgada con trombo no fatal y la posterior curación con formación de tejido fibroso y la remodelación constrictiva.
EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG |
▶ Mecanismos de ruptura de la placa
La ruptura de la placa se produce cuando la cubierta es más delgada y está más infiltrado por células espumosas (macrófagos). En las placas excéntricas, el punto más débil es a menudo el margen o región del hombro,86 y sólo las placas con cubiertas fibrosas extremadamente delgadas corren el riesgo de ruptura.
Evaluadas por examen microscópico en un estudio de autopsias de muerte cardiaca repentina, el espesor medio de las cubiertas rotas se encontró que era sólo de 23 micras y el 95% de las cubiertas fibrosos rotas estaban por debajo de 65 μm.91
Basándose en estas observaciones, Virmani et al19 introdujeron el término fibroateromas con cubierta delgada (FCFs) para los fibroateromas coronarios con un espesor de capa fibrosa <65 micras, de este modo se puede asumir que incluyen a la mayoría de las placas con riesgo de ruptura.
El adelgazamiento de la capa fibrosa implica probablemente dos mecanismos simultáneos. Uno de ellos es la pérdida gradual de las células musculares lisas (CML) de la capa fibrosa. Las cápsulas rotas contienen menos CML y menos colágeno que las cápsulas intactas,93 y las CML están generalmente ausentes en el sitio real de ruptura.93, 94 Al mismo tiempo, los macrófagos infiltrantes degradan la matriz rica en colágeno.
La capacidad de la terapia con estatinas para proteger rápidamente contra eventos coronarios indica que este tipo de inflamación también está impulsada por las lipoproteínas. Las placas rotas examinados en la autopsia están por lo general muy infiltradas por células espumosas de macrófagos,64, 94 que segregan enzimas proteolíticas tales como activadores del plasminógeno, catepsinas, y metaloproteinasas de la matriz. Las metaloproteinasas de la matriz son secretadas como zimógenos latentes que requieren de la activación extracelular, después de lo cual son capaces de degradar virtualmente todos los componentes de la matriz extracelular.
Como prueba de principio, los macrófagos que sobreexpresan una forma mutante constitutivamente activa de la matriz metaloproteinasa-9 causan la ruptura de la placa en un modelo de ratón con aterosclerosis.95 Si el adelgazamiento de la capa fibrosa toma décadas para evolucionar o es mucho más dinámico es algo que todavía no se conoce. Sin embargo, el hecho de que los fibroateromas se observen con frecuencia a partir de los 30 años de edad,17,47 donde los síndromes coronarios agudos son extremadamente raros, parece indicar que se trata de un proceso de lenta evolución.
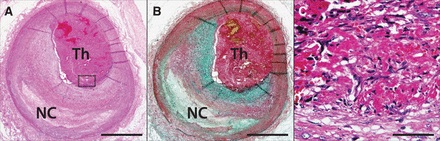
Figura 4. Erosión de la placa. A y B, la erosión de la placa con trombo oclusivo luminal (Th) y fibroateroma subyacente con núcleo necrótico temprano (NC). Una imagen de alta potencia en C muestra la invasión de células parecidas a las células del músculo liso y células endoteliales. Hematoxilina-eosina (A y C) y las manchas Pentachrome Movat (B). Las barras de escala 1 mm (A y B) y 50 micras (C). Reproducido con permiso de Kramer et al100 con permiso del editor. Copyright 2010, el American College of Cardiology Foundation.
EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG |
♦ Desencadenantes (gatillos)
La ruptura de una cubierta delgada y la posterior trombosis pueden ocurrir espontáneamente, pero en algunos casos, un aumento temporal de la tensión emocional o física proporciona la activación final del evento (gatillos).
Los disparadores reconocidos incluyen la actividad física y sexual, la ira, la ansiedad, el estrés laboral, los terremotos, la guerra, los ataques terroristas, los cambios de temperatura, las infecciones y el consumo de cocaína.96
También las actividades diarias simples o el ritmo circadiano de las vías biológicas pueden determinar la aparición de un síndrome coronario agudo (SCA), que son más frecuentes durante la mañana.97
Las vías de activación pueden incluir la activación del sistema nervioso simpático con aumento del ritmo cardíaco y la presión arterial, que lleva a la ruptura de placa, o el aumento de la coagulabilidad y de la reactividad de las plaquetas, lo que conduce a una respuesta trombótica acentuada en placas ya rotas.96-98
Es importante señalar que a pesar de que los factores desencadenantes pueden aumentar temporalmente el riesgo relativo de SCA en personas susceptibles, esto significa poco para el riesgo absoluto porque la exposición es transitoria y, a menudo relativamente infrecuente.96 Además, algunos estudios de disparadores poblacionales como el terremoto de Northridge en Los Ángeles en 1994, indican que los episodios coronarios que no se precipitaron por un disparador a menudo se habrían producido de todos modos, en ausencia de disparadores unas semanas más tarde.99
▶ Mecanismos de erosión
El mecanismo que conduce al trombo sin ruptura es una de las cuestiones sin resolver más importantes dentro de la investigación en aterosclerosis. La superficie del endotelio bajo el trombo está normalmente ausente, pero no se han identificado características morfológicas distintas de la placa subyacente. Las placas erosionadas y trombosadas que causan la muerte súbita cardiaca a menudo están apenas calcificados, a menudo están asociados con remodelación negativa, y menos inflamadas que las placas rotas.19, 100

Figura 5. Capa delgada de Fibroateroma inflamación en ca tapa. A y B, la placa está compuesta predominantemente de tejido fibroso y un núcleo necrótico (NC) con una capa fibrosa muy delgada (FC). C, tinción para CD68 (macrófagos rojos) visualiza abundantes macrófagos en el tapón fibroso, pero otras partes de la placa no se inflama. D, mayor ampliación de inserción en C. La tapa contiene pocos CML (no mostrados). Macrófagos muertos sin núcleo son vistos dentro del núcleo necrótico (*). Penetración del medio de contraste (Co) inyecta post mortem de la luz en el núcleo necrótico revela que en este caso particular una ruptura tapa está presente en un segmento adyacente. HE indica hematoxilina-eosina.
EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG |
En algunos, pero no en otros, han informado inflamación en el centro, inmediatamente debajo de la trombosis superpuesta.19, 94 El vasoespasmo se ha sugerido como una causa del daño endotelial y posterior trombosis.101 De acuerdo con esta hipótesis, las lesiones por erosión de la placa típicamente muestran láminas internas y externos intactas y elásticas y una capa media bien desarrolladas con células musculares lisas contráctiles a diferencia de las lesiones por ruptura de la placa, donde la lámina interna intacta es a menudo interrumpida y las medias subyacentes son delgadas y desorganizadas.102
Curiosamente, la morfología idéntica a la de la erosión de la placa (denudación endotelial sobre engrosamiento de la íntima patológica y fibroateromas con cubierta delgada) a menudo se pueden encontrar por encima o por debajo de las placas rotas bajo de la rotura de la placa con un trombo superpuesto fatal.19
También podría sugerir que la pérdida de endotelio puede ocurrir a veces en segundo lugar a la formación de trombos, a condición de que se supone que la ruptura vecina en estos casos es la única causa precipitante. Los estudios de autopsias indican que sólo una minoría de las rupturas conduce a los síntomas clínicos, mientras que las otras se curan en silencio con sólo un trombo mural.103-105 Hipotéticamente, la pérdida de las propiedades antitrombóticas de la superficie de la placa, lo que se puede presentar en la erosión como placa, podría ser un factor determinante entre estos resultados, junto con la circulación factores trombogénicos.
▶ Trombosis
La magnitud de la respuesta trombótica en las placas rotas o erosionadas es extremadamente variable, y sólo de vez en cuando se produce un trombo luminal importante y potencialmente mortal. Probablemente los factores determinantes son los de la triada clásica de Virchow: (1) la trombogenicidad del material de la placa expuesta, (2) las perturbaciones de flujo locales, y (3) propensión trombótica sistémica. El tabaquismo predispone a las enfermedades del corazón, al menos en parte, a través del aumento propensión trombótica sistémica.96, 106
Con la ruptura de placa, la cubierta de colágeno y el núcleo lipídico altamente trombogénico, enriquecido con factor tisular que expresan micropartículas apoptóticas, están expuestos a los factores trombogénicos de la sangre.107, 108 La erosión placa es probablemente un estímulo trombogénico más débil y, por lo tanto, otros factores de la tríada puede ser particularmente importantes en este contexto. Consistentemente, los trombos fatales asociados con erosión de la placa parecen ser más largos que los que se producen por la ruptura de la placa.100
La relación entre el tiempo de ruptura de la placa y el síndrome de inicio no se evalúa con facilidad debido a que la ruptura en sí misma es asintomática y el siguiente proceso trombótico es altamente impredecible. El material de la placa se encuentra a veces intercalado en el trombo,64,94 lo que indica que la trombosis severa es seguida inmediatamente después de la ruptura de placa. En otros casos, la respuesta trombótica es dinámica: la trombosis y la trombólisis, a menudo asociadas con vasoespasmo, tienden a ocurrir simultáneamente causando alteraciones del flujo intermitente y la formación de un trombo en capas que se desarrolla durante días.109
Los trombos obtenidos mediante trombectomía o postmortem analizados pueden ser parcialmente degradados u organizados, lo que indica que se iniciaron o bien varios días o hasta 1 semana antes de la presentación clínica del SCA.100, 110 Es concebible que el trombo durante este período, como en la inflamación inducida por la trombosis de las venas111 podría dar lugar a desprendimiento endotelial e infiltración de neutrófilos y monocitos / macrófagos en la pared vascular subyacente, lo que podría suponerse equivocadamente que existían antes de la iniciación del trombo.
Aunque el flujo de sangre continúa a través de la lesión culpable, las microembolias de material de la placa y los trombos pueden ser lavados, lo que conduce a la embolización distal de la miocardio.109-112 Se han reportado tales émbolos como más frecuentes en las erosiones (74%) que en las rupturas (40%) en individuos que presentan muerte coronaria repentina en ausencia de cualquier intervención coronaria,113 que en sí misma puede producir embolización iatrogénica.112 La embolia distal de cualquier fuente puede causar obstrucción microvascular que evita la perfusión miocárdica a pesar de que la arteria coronaria relacionada con el infarto se haya recanalizado.
▶ Placas curadas y trombos incorporadosUn patrón no uniforme especial de tipo denso de tipo I (mayores) y laxo III (menores) de colágeno, juzgada para indicar ruptura de la placa curada, puede ser identificado en muchas placas coronarias, especialmente en aquellos que causan la estenosis crónica de alto grado (Figura 6).104 a menudo varios sitios de ruptura curadas están presentes, y el número de rupturas curadas se correlaciona con la gravedad de stenosis.105 Otras placas pueden tener una apariencia de múltiples capas consistente con una historia de incorporación y organización de trombo en plaques.19 erosionados Aunque estos resultados son observacional, indican que la placa se rompe en silencio y erosiones son importantes para el crecimiento de la placa y la formación de estenosis crónica (Figura 3). Por otra parte, similar a una cicatriz contracción del tejido fibroso de cicatrización se ha propuesto como una causa de la remodelación constrictiva menudo visto con plaques.84 severamente estenótica,114 Un papel central de la curación de la placa en la formación de estenosis puede explicar por qué la estenosis coronaria crónica a menudo se desarrolla en un lugar fásica de manera lineal, formando en los sitios que sólo se redujeron de manera insignificante en un angiografía.104, 115

Figura 6. Sanado ruptura de la placa. A, fibroateroma con hemorragia en un núcleo necrótico tarde (NC) se ve que subyace un trombo sanado (HTH). Movat pentacromo mancha. B, mayor aumento de la trombo curado muestra células de músculo liso extensas dentro de una neoíntima proteoglicanos ricos en colágeno y demarcación clara de la región de la placa fibrosa a la derecha. C, Las capas de colágeno se muestra por tinción con rojo Sirius. Tenga en cuenta el área de densa colágeno rojo, oscuro que rodea el núcleo necrótico presumiblemente corresponde a la vieja capa fibrosa. D, de imagen de la misma sección tomada con luz polarizada. Colágeno denso (tipo I) en la vieja capa fibrosa es rojizo más ligero de color amarillo y se interrumpe (flecha), con los nuevos tipo verdoso colágeno III en la derecha y por encima de zona de rotura. Reproducido con permiso de Burke et al105 con permiso del editor. Copyright 2001, Asociación Americana del Corazón.
EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG |
▶ Carga de placas
En la investigación y en la práctica clínica, existe una necesidad de caracterizar la aterosclerosis o a las placas individuales por algunas características importantes y medibles que indican el estado del proceso de la enfermedad y el riesgo de progresión. Estas variables se pueden utilizar como puntos finales para los ensayos clínicos y como herramientas de predicción del riesgo para orientar las decisiones acerca de las terapias. Algunos de los términos utilizados en esta área son: carga de placas, la actividad y vulnerabilidad.
La carga de placa es una medida de la extensión de la aterosclerosis en el cuerpo o en un lecho vascular particular, independientemente de la composición y la actividad de las placas celulares. Se puede medir como volumen de la placa, la superficie cubierta con lesiones arteriales, o por algún representante correlacionado, tales como la medición del calcio coronario mediante tomografía computarizada, el grosor íntima-media y el área de la placa en las carótidas por ultrasonido, y el índice de presión tobillo-braquial en la enfermedad vascular periférica.
Debido a que la aterosclerosis es una enfermedad multifocal que afecta a toda la vasculatura, tiene una alta carga de placa en un territorio, por ejemplo, en las carótidas o en las extremidades inferiores, también puede ser un marcador para la enfermedad avanzada en otros lugares, especialmente en las arterias coronarias debido a su alta susceptibilidad.47, 116-118 Así, la presencia de una estenosis carotídea detectada por ecografía o la enfermedad vascular periférica detectada por un bajo índice tobillo-brazo lleva un riesgo de IAM comparable al de los pacientes con enfermedad coronaria y por lo tanto se tratan como equivalentes del riesgo de cardiopatía coronaria.119
Un razonamiento similar se aplica a la perspectiva de encontrar biomarcadores plasmáticos correlacionados con la aterosclerosis. Debido a que la mayor parte de la aterosclerosis está situada en la aorta abdominal y en las arterias iliofemorales, se esperaría que cualquier biomarcador del plasma de la aterosclerosis para reflejar la extensión de la aterosclerosis en esos territorios, a continuación, y por un argumento similar al anterior indicaría el riesgo de tener aterosclerosis coronaria concomitante.
En consonancia con que la placa está compuesta predominantemente por tejido fibrótico hipocelular con baja renovación celular, 68.120 la cantidad de placa cambia muy modesta lentamente, incluso con el tratamiento que reduce sustancialmente el riesgo de SCA.121 De los diferentes componentes de las lesiones ateroscleróticas progresivas, los lípidos y los macrófagos parecen ser los que más se prestan para la regresión.
▶ Actividad de la placa
La actividad de la enfermedad o de las placas individuales es un importante, pero difícil, concepto. Es importante porque la capacidad de medir la actividad de la enfermedad con fidelidad, por ejemplo, por proyección de imágenes no invasiva o mediante un biomarcador circulante en el plasma, sería una herramienta importante para el descubrimiento de los factores causales y para demostrar la eficacia de los tratamientos en ensayos clínicos pequeños.
Por otra parte, permitiría no tener que depender de los puntos finales clínicos para medir el efecto que allanaría el camino para la investigación y, posiblemente, el tratamiento preventivo en las primeras etapas de la enfermedad, donde parecemos saber más acerca de los procesos fisiopatológicos y donde la enfermedad puede ser potencialmente más modificable.
Se trata de un concepto difícil porque la actividad de la enfermedad o de la placa no tiene un significado simple y definido. A menudo se considera que significa inflamación, medida, por ejemplo como la densidad de macrófagos en las placas. Esto es razonable dado el papel central de la inflamación vascular en el desarrollo de la placa. Sin embargo, hay una diferencia fundamental entre la inflamación de una lesión aterosclerótica temprana y la inflamación focal de una capa fibrosa que puede conducir a la ruptura y trombosis.124
Varios otros procesos en las placas ateroscleróticas podrían incluirse bajo el título de actividad de la placa, incluyendo la necrosis de la íntima, que constituye la actividad quizás más perjudicial de la enfermedad. La angiogénesis, la permeabilidad del endotelio, y el sangrado o hemorragia de la placa a menudo acompañan a la inflamación y constituyen otros biomarcadores potenciales de la actividad de la enfermedad.1
Para predecir qué placas se encuentran en riesgo de precipitar la trombosis y comprender los mecanismos que conducen a su formación, se ha puesto mucho esfuerzo en el reconocimiento de las características patológicas de las placas vulnerables (o placas trombogénicas o de alto riesgo): con alto riesgo a corto plazo de trombosis.87
En particular, la presencia de trombosis no es lo misma que la aparición de un síndrome coronario agudo. Como se ha expuesto en los apartados anteriores, muchos, quizá la mayoría, de las roturas y erosiones de las placas son asintomáticas en el corto plazo, aunque a veces pueden conducir a la obstrucción gradual del vaso coronaria en el paciente vulnerable que es un término usado para describir a los pacientes en alto a corto riesgo a largo plazo de padecer un evento clínico agudo Esto depende de la carga de placa, de la vulnerabilidad de la placa, de la propensión trombótica sistémica y de la susceptibilidad del miocardio a la isquemia y a la arritmia.
La placa vulnerable se utiliza a veces como un concepto que comprende a las placas de alto riesgo de trombosis por cualquier mecanismo (rotura, erosión) y, a veces para describir un conjunto de características histológicas que por asociación se supone que aumentará el riesgo de ruptura inminente y trombosis. El uso de este último explica por qué se utiliza también el término placa vulnerable en la investigación con modelos de ratón, donde la ocurrencia de trombosis en placas es extremadamente rara.
En la tabla se describen las funciones que se han encontrado para caracterizar a las placas rotas en estudios de autopsia. Por inferencia, las mismas características, excepto el trombo y la ruptura de la cápsula, se supone que son útiles para marcar a las placas vulnerables (tendencia a la rotura).
Tabla. Características de la ruptura de placas *
1. Trombo
2. Núcleo necrótico grande
3. Capa fibrosa que recubre el núcleo necrótico
4. Remodelación expansiva preservando el lumen
5. Neovascularización desde los vasa vasorum
6. Hemorragia de la placa
7. Adventicia / inflamación perivascular
8. Calcificación granular
EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG
|
La placa prototípica con tendencia a la ruptura es un gran fibroateroma con una cubierta delgada, un gran núcleo y con infiltración de macrófagos.86, 93 Aunque la densidad de macrófagos en las cubiertas rotas es generalmente alta,93 la densidad de macrófagos en toda la placa rara vez supera un pequeño porcentaje porque las cubiertas rotas son pequeñas,124 y, por lo tanto, es un error pensar que las placas que se rompen siempre están muy inflamadas.
Otras características asociadas incluyen tamaño de la placa grande, la remodelación expansiva que atenúa la obstrucción luminal (estenosis leve por angiografía), la neovascularización,la hemorragia de la placa, la inflamación de la adventicia, y un patrón irregular de calcificaciones.124
Las cubiertas delgadas de las placas son los posibles precursores de la mayoría de las rupturas.88, 91 Tienden a agruparse en los segmentos proximales de las arterias coronarias, donde se ven la mayoría de las placas que se rompen y los trombo.126 Rara vez se ven más de unas pocos cubiertas delgadas simultáneamente.127, 128
La ausencia de cubiertas delgadas en un paciente indica un riesgo inminente bajo de ruptura de la placa y de trombosis. Un intento de medir el riesgo conferido por su presencia se hizo en reciente estudio PROSPECT (Observaciones regionales para estudiar predictores de eventos en el árbol coronario). De 595 análisis de histología virtual se identificaron placas con cubiertas delgadas en 313 de 623 pacientes examinados mediante ecografía intravascular, sólo 26 llevaron a eventos coronarios (angina progresiva) en una mediana de seguimiento de 3.4 años.129
Otra serie reciente serie estudio intravascular por ultrasonido reveló que las cubiertas delgadas surgen y desaparecen de forma más dinámica de lo que se pensaba anteriormente.130 Ambos estudios debilitan los fundamentos de un enfoque terapéutico dirigido a las placas propensas a la ruptura. Sin embargo, es importante señalar que el rendimiento y la resolución de la ecografía intravascular no permite la identificación de cubiertas delgadas definidas como lo hacen los anatomopatólogos131, 132 y el uso de métodos más precisos en futuros estudios puede conducir a diferentes resultados.
Si no hay ningún núcleo necrótico, no hay capa fibrosa que recubra la ruptura. Consistentemente, el no tener núcleos necróticos entre las lesiones no responsables de las arterias coronarias proximales indica un pronóstico favorable después de un síndrome coronario agudo.133 Sin embargo, un núcleo necrótico más grande también confiere un riesgo mayor que uno pequeño.101,134
La importancia del tamaño del núcleo necrótico para la estabilidad de la placa es comprensible porque la expansión del núcleo puede erosionar la capa fibrosa desde abajo y debido a la falta total de apoyo de colágeno en el núcleo rico en lípidos confiere una mayor tensión de tracción a la capa fibrosa que lo recubre. Un núcleo necrótico grande también puede aumentar la trombogenicidad del material de la placa y por lo tanto el riesgo de un evento clínico en caso de ruptura de placa.107
▶ Tamaño de la placa y severidad de la estenosis
Estudios angiográficos retrospectivos y recientemente el estudio PROSPECT han demostrado que las placas que causan estenosis tienen un mayor riesgo de producir un evento clínico que las placas que no lo hacen.86, 129 Sin embargo, la mayoría de los síndromes coronarios agudos se precipitan por placas que no causaban una estenosis significativa en la angiografía semanas o meses antes, simplemente porque estas placas son mucho más frecuentes que las pocas placas estenóticas, incluso si tienen una menor vulnerabilidad.86, 129 La estenosis preexistente leve en estos casos se debe a la remodelación expansiva debido a que placas rotas son en general grandes.135, 136
Estudios recientes han descrito estenosis angiográfica significativa en los días previos a un infarto de miocardio.137,138 Por otra parte, los datos de autopsia muestran que las cubiertas fibrosas delgadas son más pequeñas que las placas rotas, y éstas son casi siempre grandes y parecen gravemente obstructivas cuando se estudian bajo el microscopio.139 Estas observaciones han cuestionado la noción largamente sostenida de que las lesiones obstructivas coronarias leves a moderadas son responsables de la mayoría de los infarto de miocardio.13, 137
Sin embargo, puede no ser una sorpresa que un examen angiográfico realizado cerca de un IAM revele una lesión coronaria grave o que el tamaño de una placa rota supere a la de su precursora. La ruptura de la placa es seguido no sólo por una trombosis luminal dinámica (± vasoespasmo), sino también por la hemorragia desde la luz a la placa a través de la superficie de ruptura (hemorragia en el núcleo necrótico que pueden haber precedido a la ruptura),88 lo que dio lugar a la expansión rápida de la placa rápida que no responde a la trombólisis ni a la trombectomía por aspiración, y la relación temporal entre la ruptura de la placa y el SCA es a menudo posterior.110
Además, es importante tener en cuenta que el área transversal de la estenosis determinada a partir de las lesiones, incluidas en parafina y fijadas en formol no equivale a diámetro luminal de la estenosis determinada por angiografía, debido a la remodelación arterial, la conversión matemática entre el diámetro y el área, y a la contracción del tejido de fijación y deshidratación.135

Figura 7. Evaluaciones angiográficas y en sección transversal de la luz arterial. Fila superior, angiográficas. Los ejemplos (A-D) ilustran cómo la angiografía no se puede utilizar para determinar la presencia o tamaño de las placas ateroscleróticas por la remodelación arterial. Fila inferior, vistas en sección transversal. Los mismos segmentos visto bajo el microscopio que ilustra cómo la estenosis del lumen no se puede determinar debido a la remodelación arterial. Una estenosis angiográfica ≤ 50% puede parecer gravemente obstruida. Fibroateromas Thin-de cabeza y las placas rotas suelen ser grandes y se asocia con la remodelación expansiva, como se muestra en el ejemplo D. Reproducido con permiso de Fishbein y Siegel135 con permiso del editor. Derechos reservados 1996, la Asociación Americana del Corazón.
EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG |
▶ Otras características asociadas a la ruptura de la placa
Existen ciertas características adicionales de la placa son más comunes en las placas rotas que en las placas vírgenes, entre ellas el aumento de la neovascularización y la inflamación adventicia.140, 141 Por otra parte, las lesiones responsables de síndromes coronarios agudos son generalmente menos calcificadas que las placas responsables de la angina de pecho estable y el patrón de calcificación de la placa también difiere.80, 142 Estas características no están asociadas de forma independiente con la ruptura de la placa, sin embargo, y si existe una relación causal, es probable que sea a través de la modulación de la inflamación o del espesor de la capa fibrosa o del tamaño del núcleo necrótico. La importancia de estas características, sin embargo, radica en el hecho de que pueden ser dianas para la ontención de imágenes no invasivas.
La mayor parte de nuestro conocimiento mecanicista de la aterosclerosis se relaciona con el desarrollo de placas ateroscleróticas, incluyendo la influencia de las LDL en la inflamación vascular, pero la prevención primaria dirigida rara vez se inicia antes de que las primeros complicaciones clínicas de la aterosclerosis tengan lugar.143 En esta etapa de la enfermedad, se sabe comparativamente poco acerca de los procesos biológicos relevantes contra los cuales puede ser posible intervenir.
El conocimiento de las causas del adelgazamiento de la cápsula fibrosa y de la ruptura de la placa es incompleto, y existen grandes lagunas en nuestra comprensión de los mecanismos que llevan a la trombosis de las placas sin ruptura. Además, las placas en pacientes con SCA son relativamente hipocelulares e inertes y con baja rotación de los tejidos, lo que puede disminuir por completo la posibilidad de cambiar su destino por la intervención farmacológica.68, 120
Las mediciones de la carga de placa en las personas de mediana edad pueden mejorar la estratificación de riesgo que permita el tratamiento preventivo que se inidicará a más personas en riesgo.144 Sin embargo, en la búsqueda de mejoras en las estrategias de prevención individualizadas, no se debe olvidar la importancia continua de los enfoques de salud pública,145 tales como la promoción de la calidad de la dieta, dejar de fumar, la actividad física y el control de peso a través de la información y de la legislación.
Durante las últimas décadas, los niveles de los factores de riesgo han disminuido sustancialmente en muchos países desarrollados, en especial para el tabaquismo y el colesterol y la carga de placa entre los jóvenes miembros del servicio de Estados Unidos es ahora al parecer sólo una fracción de lo que fue durante la guerra de Corea.146
La calidad de las iniciativas de salud pública, sin embargo, podrían mejorarse considerablemente mediante el desarrollo de técnicas no invasivas para medir la actividad de la enfermedad aterosclerótica en las personas asintomáticas lo que permite recomendaciones sobre los hábitos del estilo de vida y su incorporación a los estudios clínicos en lugar de hacerlo solo con las asociaciones epidemiológicas.
►Artículo ➔ Noticia ➲ Tema básico ➜ Editorial
► Infarto Agudo de Miocardio
➲ Infarto agudo del miocardio
➲ Síndromes coronarios agudos
➲ El Electrocardiograma (EKG,ECG)
► Un extraño caso de infarto agudo del miocardio
➲ Palpitaciones: causa de consultas en salas de Emergencia
➲ Evaluación y clasificación de los pacientes con lesión e infarto del miocardio
► Reconocimiento de síntomas en mujeres jóvenes con infarto agudo del miocardio
► Estatinas en pacientes con síndromes coronarios agudos sometidos a angioplastia
► Administración prehospitalaria de ticagrelol en pacientes con infarto agudo de miocardio con ST elevado
➲ Infarto agudo del miocardio
➲ Síndromes coronarios agudos
➲ El Electrocardiograma (EKG,ECG)
► Un extraño caso de infarto agudo del miocardio
➲ Palpitaciones: causa de consultas en salas de Emergencia
➲ Evaluación y clasificación de los pacientes con lesión e infarto del miocardio
► Reconocimiento de síntomas en mujeres jóvenes con infarto agudo del miocardio
► Estatinas en pacientes con síndromes coronarios agudos sometidos a angioplastia
► Administración prehospitalaria de ticagrelol en pacientes con infarto agudo de miocardio con ST elevado
1.↵ Levy D. Combating the epidemic of heart disease. JAMA. 2012;308:2624–2625. CrossRef Search Google Scholar
2.↵ Murray CJ, Lopez AD. Measuring the global burden of disease. N Engl J Med. 2013;369:448–457. CrossRefMedline Search Google Scholar
3.↵ Laslett LJ, Alagona P Jr., Clark BA III., Drozda JP Jr., Saldivar F, Wilson SR, Poe C, Hart M. The worldwide environment of cardiovascular disease: prevalence, diagnosis, therapy, and policy issues: a report from the American College of Cardiology. J Am Coll Cardiol. 2012;60:S1–S49. CrossRefMedline Search Google Scholar
4.↵ Fuster V, Mearns BM. The CVD paradox: mortality vs prevalence. Nat Rev Cardiol. 2009;6:669. CrossRefMedline Search Google Scholar
5.↵ Nabel EG, Braunwald E. A tale of coronary artery disease and myocardial infarction. N Engl J Med. 2012;366:54–63. CrossRefMedline Search Google Scholar
6.↵ Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A, Lanas F, McQueen M, Budaj A, Pais P, Varigos J, Lisheng L; INTERHEART Study Investigators. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet. 2004;364:937–952. CrossRefMedline Search Google Scholar
7.↵ Lim SS, Vos T, Flaxman AD, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2224–2260. CrossRefMedline Search Google Scholar
8.↵ Steinberg D, Glass CK, Witztum JL. Evidence mandating earlier and more aggressive treatment of hypercholesterolemia. Circulation. 2008;118:672–677. FREE Full Text
9.↵ Ference BA, Yoo W, Alesh I, Mahajan N, Mirowska KK, Mewada A, Kahn J, Afonso L, Williams KA, Flack JM. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease. J Am Coll Cardiol. 2012;60:2631–2639. CrossRefMedline Search Google Scholar
10.↵ Bøttcher M, Falk E. Pathology of the coronary arteries in smokers and non-smokers. J Cardiovasc Risk. 1999;6:299–302. Medline Search Google Scholar
11.↵ Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics–2014 update: a report from the American Heart Association. Circulation. 2014;129:e28–e292. FREE Full Text
12.↵ Fowkes FG, Rudan D, Rudan I, Aboyans V, Denenberg JO, McDermott MM, Norman PE, Sampson UK, Williams LJ, Mensah GA, Criqui MH. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: a systematic review and analysis. Lancet. 2013;382:1329–1340. CrossRefMedline Search Google Scholar
13.↵ Willey J, Gonzalez-Castellon M. Cholesterol level and stroke: a complex relationship. JAMA Intern Med. 2013;173:1765–1766. CrossRef Search Google Scholar
14.↵ Bentzon JF, Falk E. Atherosclerotic lesions in mouse and man: is it the same disease? Curr Opin Lipidol. 2010;21:434–440. CrossRefMedline Search Google Scholar
15.↵ Stary HC, Blankenhorn DH, Chandler AB, Glagov S, Insull W Jr., Richardson M, Rosenfeld ME, Schaffer SA, Schwartz CJ, Wagner WD. A definition of the intima of human arteries and of its atherosclerosis-prone regions. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1992;85:391–405. FREE Full Text
16.↵ Stary HC, Chandler AB, Glagov S, Guyton JR, Insull W Jr., Rosenfeld ME, Schaffer SA, Schwartz CJ, Wagner WD, Wissler RW. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler Thromb. 1994;14:840–856. Abstract/FREE Full Text
17.↵ Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W Jr., Rosenfeld ME, Schwartz CJ, Wagner WD, Wissler RW. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1995;92:1355–1374. Abstract/FREE Full Text
18.↵ Stary HC. Natural history and histological classification of atherosclerotic lesions: an update. Arterioscler Thromb Vasc Biol. 2000;20:1177–1178. FREE Full Text
19.↵ Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20:1262–1275. FREE Full Text
20.↵ Wentzel JJ, Chatzizisis YS, Gijsen FJ, Giannoglou GD, Feldman CL, Stone PH. Endothelial shear stress in the evolution of coronary atherosclerotic plaque and vascular remodelling: current understanding and remaining questions. Cardiovasc Res. 2012;96:234–243. Abstract/FREE Full Text
21.↵ Millonig G, Niederegger H, Rabl W, Hochleitner BW, Hoefer D, Romani N, Wick G. Network of vascular-associated dendritic cells in intima of healthy young individuals. Arterioscler Thromb Vasc Biol. 2001;21:503–508. Abstract/FREE Full Text
22.↵ Schwartz SM, deBlois D, O’Brien ER. The intima. Soil for atherosclerosis and restenosis. Circ Res. 1995;77:445–465. FREE Full Text
23.↵ Velican D, Velican C. Atherosclerotic involvement of the coronary arteries of adolescents and young adults. Atherosclerosis. 1980;36:449–460. CrossRefMedline Search Google Scholar
24.↵ Roberts WC. Coronary atherosclerosis: is the process focal or diffuse among patients with symptomatic or fatal myocardial ischemia? Am J Cardiol. 1998;82:41T–44T. Medline Search Google Scholar
25.↵ Cheng C, Tempel D, van Haperen R, van der Baan A, Grosveld F, Daemen MJ, Krams R, de Crom R. Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation. 2006;113:2744–2753. Abstract/FREE Full Text
26.↵ Steinberg D, Witztum JL. Oxidized low-density lipoprotein and atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:2311–2316. FREE Full Text
27.↵ Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. CrossRefMedline Search Google Scholar
28.↵ Subramanian M, Tabas I. Dendritic cells in atherosclerosis. Semin Immunopathol. 2014;36:93–102. CrossRef Search Google Scholar
29.↵ Skålén K, Gustafsson M, Rydberg EK, Hultén LM, Wiklund O, Innerarity TL, Borén J. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. 2002;417:750–754. CrossRefMedline Search Google Scholar
30.↵ Tabas I, Williams KJ, Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116:1832–1844. Abstract/FREE Full Text
31.↵ Stamler J, Daviglus ML, Garside DB, Dyer AR, Greenland P, Neaton JD. Relationship of baseline serum cholesterol levels in 3 large cohorts of younger men to long-term coronary, cardiovascular, and all-cause mortality and to longevity. JAMA. 2000;284:311–318. CrossRefMedline Search Google Scholar
32.↵ Hartiala O, Magnussen CG, Kajander S, et al. Adolescence risk factors are predictive of coronary artery calcification at middle age: the cardiovascular risk in young Finns study. J Am Coll Cardiol. 2012;60:1364–1370. CrossRefMedline Search Google Scholar
33.↵ Leitinger N, Schulman IG. Phenotypic polarization of macrophages in atherosclerosis. Arterioscler Thromb Vasc Biol. 2013;33:1120–1126. Abstract/FREE Full Text
34.↵ Bouhlel MA, Derudas B, Rigamonti E, Dièvart R, Brozek J, Haulon S, Zawadzki C, Jude B, Torpier G, Marx N, Staels B, Chinetti-Gbaguidi G. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–143. CrossRefMedline Search Google Scholar
35.↵ Stöger JL, Gijbels MJ, van der Velden S, Manca M, van der Loos CM, Biessen EA, Daemen MJ, Lutgens E, de Winther MP. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis. 2012;225:461–468. CrossRefMedline Search Google Scholar
36.↵ Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10:36–46. CrossRefMedline Search Google Scholar
37.↵ Hansson GK, Jonasson L. The discovery of cellular immunity in the atherosclerotic plaque. Arterioscler Thromb Vasc Biol. 2009;29:1714–1717. Abstract/FREE Full Text
38.↵ Hansson GK, Nilsson J. Vaccination against atherosclerosis? Induction of atheroprotective immunity. Semin Immunopathol. 2009;31:95–101. CrossRefMedline Search Google Scholar
39.↵ Steinberg D. The LDL modification hypothesis of atherogenesis: an update. J Lipid Res. 2009;50(suppl):S376–S381. Abstract/FREE Full Text
40.↵ Kunjathoor VV, Febbraio M, Podrez EA, Moore KJ, Andersson L, Koehn S, Rhee JS, Silverstein R, Hoff HF, Freeman MW. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem. 2002;277:49982–49988. Abstract/FREE Full Text
41.↵ Haka AS, Grosheva I, Chiang E, Buxbaum AR, Baird BA, Pierini LM, Maxfield FR. Macrophages create an acidic extracellular hydrolytic compartment to digest aggregated lipoproteins. Mol Biol Cell. 2009;20:4932–4940. Abstract/FREE Full Text
42.↵ Witztum JL. You are right too! J Clin Invest. 2005;115:2072–2075. CrossRefMedline Search Google Scholar
43.↵ Katsuda S, Boyd HC, Fligner C, Ross R, Gown AM. Human atherosclerosis. III. Immunocytochemical analysis of the cell composition of lesions of young adults. Am J Pathol. 1992;140:907–914. Medline Search Google Scholar
44.↵ Napoli C, D’Armiento FP, Mancini FP, Postiglione A, Witztum JL, Palumbo G, Palinski W. Fatty streak formation occurs in human fetal aortas and is greatly enhanced by maternal hypercholesterolemia. Intimal accumulation of low density lipoprotein and its oxidation precede monocyte recruitment into early atherosclerotic lesions. J Clin Invest. 1997;100:2680–2690. CrossRefMedline Search Google Scholar
45.↵ Stary HC. Lipid and macrophage accumulations in arteries of children and the development of atherosclerosis. Am J Clin Nutr. 2000;72:1297S–1306S. Abstract/FREE Full Text
46.↵ Kolodgie FD, Burke AP, Nakazawa G, Virmani R. Is pathologic intimal thickening the key to understanding early plaque progression in human atherosclerotic disease? Arterioscler Thromb Vasc Biol. 2007;27:986–989. FREE Full Text
47.↵ Dalager S, Paaske WP, Kristensen IB, Laurberg JM, Falk E. Artery-related differences in atherosclerosis expression: implications for atherogenesis and dynamics in intima-media thickness. Stroke. 2007;38:2698–2705. Abstract/FREE Full Text
48.↵ Kolodgie FD, Gold HK, Burke AP, Fowler DR, Kruth HS, Weber DK, Farb A, Guerrero LJ, Hayase M, Kutys R, Narula J, Finn AV, Virmani R. Intraplaque hemorrhage and progression of coronary atheroma. N Engl J Med. 2003;349:2316–2325. CrossRefMedline Search Google Scholar
49.↵ Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. CrossRefMedline Search Google Scholar
50.↵ Clarke MC, Bennett MR. Cause or consequence: what does macrophage apoptosis do in atherosclerosis? Arterioscler Thromb Vasc Biol. 2009;29:153–155. FREE Full Text
51.↵ Myoishi M, Hao H, Minamino T, Watanabe K, Nishihira K, Hatakeyama K, Asada Y, Okada K, Ishibashi-Ueda H, Gabbiani G, Bochaton-Piallat ML, Mochizuki N, Kitakaze M. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation. 2007;116:1226–1233. Abstract/FREE Full Text
52.↵ Seimon TA, Nadolski MJ, Liao X, Magallon J, Nguyen M, Feric NT, Koschinsky ML, Harkewicz R, Witztum JL, Tsimikas S, Golenbock D, Moore KJ, Tabas I. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. 2010;12:467–482. CrossRefMedline Search Google Scholar
53.↵ Lutgens E, de Muinck ED, Kitslaar PJ, Tordoir JH, Wellens HJ, Daemen MJ. Biphasic pattern of cell turnover characterizes the progression from fatty streaks to ruptured human atherosclerotic plaques. Cardiovasc Res. 1999;41:473–479. Abstract/FREE Full Text
54.↵ Crisby M, Kallin B, Thyberg J, Zhivotovsky B, Orrenius S, Kostulas V, Nilsson J. Cell death in human atherosclerotic plaques involves both oncosis and apoptosis. Atherosclerosis. 1997;130:17–27. CrossRefMedline Search Google Scholar
55.↵ Schrijvers DM, De Meyer GR, Kockx MM, Herman AG, Martinet W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:1256–1261. Abstract/FREE Full Text
56.↵ Gautier EL, Huby T, Witztum JL, Ouzilleau B, Miller ER, Saint-Charles F, Aucouturier P, Chapman MJ, Lesnik P. Macrophage apoptosis exerts divergent effects on atherogenesis as a function of lesion stage. Circulation. 2009;119:1795–1804. Abstract/FREE Full Text
57.↵ Clarke MC, Figg N, Maguire JJ, Davenport AP, Goddard M, Littlewood TD, Bennett MR. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med. 2006;12:1075–1080. CrossRefMedline Search Google Scholar
58.↵ Guyton JR. Phospholipid hydrolytic enzymes in a ‘cesspool’ of arterial intimal lipoproteins: a mechanism for atherogenic lipid accumulation. Arterioscler Thromb Vasc Biol. 2001;21:884–886. FREE Full Text
59.↵ Haka AS, Grosheva I, Singh RK, Maxfield FR. Plasmin promotes foam cell formation by increasing macrophage catabolism of aggregated low-density lipoprotein. Arterioscler Thromb Vasc Biol. 2013;33:1768–1778. Abstract/FREE Full Text
60.↵ McGill HC Jr., McMahan CA, Malcom GT, Oalmann MC, Strong JP. Effects of serum lipoproteins and smoking on atherosclerosis in young men and women. The PDAY Research Group. Pathobiological Determinants of Atherosclerosis in Youth. Arterioscler Thromb Vasc Biol. 1997;17:95–106. Abstract/FREE Full Text
61.↵ Kumamoto M, Nakashima Y, Sueishi K. Intimal neovascularization in human coronary atherosclerosis: its origin and pathophysiological significance. Hum Pathol. 1995;26:450–456. CrossRefMedline Search Google Scholar
62.↵ Sluimer JC, Kolodgie FD, Bijnens AP, Maxfield K, Pacheco E, Kutys B, Duimel H, Frederik PM, van Hinsbergh VW, Virmani R, Daemen MJ. Thin-walled microvessels in human coronary atherosclerotic plaques show incomplete endothelial junctions relevance of compromised structural integrity for intraplaque microvascular leakage. J Am Coll Cardiol. 2009;53:1517–1527. CrossRefMedline Search Google Scholar
63.↵ Davies MJ, Thomas A. Thrombosis and acute coronary-artery lesions in sudden cardiac ischemic death. N Engl J Med. 1984;310:1137–1140. CrossRefMedline Search Google Scholar
64.↵ Falk E. Plaque rupture with severe pre-existing stenosis precipitating coronary thrombosis. Characteristics of coronary atherosclerotic plaques underlying fatal occlusive thrombi. Br Heart J. 1983;50:127–134. Abstract/FREE Full Text
65.↵ Finn AV, Nakano M, Polavarapu R, Karmali V, Saeed O, Zhao X, Yazdani S, Otsuka F, Davis T, Habib A, Narula J, Kolodgie FD, Virmani R. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J Am Coll Cardiol. 2012;59:166–177. CrossRefMedline Search Google Scholar
66.↵ Purushothaman M, Krishnan P, Purushothaman KR, Baber U, Tarricone A, Perez JS, Wiley J, Kini A, Sharma SK, Fuster V, Moreno PR. Genotype-dependent impairment of hemoglobin clearance increases oxidative and inflammatory response in human diabetic atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2769–2775. Abstract/FREE Full Text
67.↵ Cahill LE, Levy AP, Chiuve SE, Jensen MK, Wang H, Shara NM, Blum S, Howard BV, Pai JK, Mukamal KJ, Rexrode KM, Rimm EB. Haptoglobin genotype is a consistent marker of coronary heart disease risk among individuals with elevated glycosylated hemoglobin. J Am Coll Cardiol. 2013;61:728–737. CrossRef Search Google Scholar
68.↵ Kragel AH, Reddy SG, Wittes JT, Roberts WC. Morphometric analysis of the composition of atherosclerotic plaques in the four major epicardial coronary arteries in acute myocardial infarction and in sudden coronary death. Circulation. 1989;80:1747–1756. Abstract/FREE Full Text
69.↵ Stary HC. An Atlas of Atherosclerosis: Progression and Regression. New York: Parthenon Publishing Group; 1999. Search Google Scholar
70.↵ Gomez D, Shankman LS, Nguyen AT, Owens GK. Detection of histone modifications at specific gene loci in single cells in histological sections. Nat Methods. 2013;10:171–177. CrossRefMedline Search Google Scholar
71.↵ Bentzon JF, Weile C, Sondergaard CS, Hindkjaer J, Kassem M, Falk E. Smooth muscle cells in atherosclerosis originate from the local vessel wall and not circulating progenitor cells in ApoE knockout mice. Arterioscler Thromb Vasc Biol. 2006;26:2696–2702. Abstract/FREE Full Text
72.↵ Campbell JH, Campbell GR. Smooth muscle phenotypic modulation—a personal experience. Arterioscler Thromb Vasc Biol. 2012;32:1784–1789. Abstract/FREE Full Text
73.↵ Feil S, Hofmann F, Feil R. SM22alpha modulates vascular smooth muscle cell phenotype during atherogenesis. Circ Res. 2004;94:863–865. Abstract/FREE Full Text
74.↵ Tang Z, Wang A, Yuan F, Yan Z, Liu B, Chu JS, Helms JA, Li S. Differentiation of multipotent vascular stem cells contributes to vascular diseases. Nat Commun. 2012;3:875. CrossRefMedline Search Google Scholar
75.↵ Bentzon JF, Falk E. Circulating smooth muscle progenitor cells in atherosclerosis and plaque rupture: current perspective and methods of analysis. Vascul Pharmacol. 2010;52:11–20. CrossRefMedline Search Google Scholar
76.↵ Nguyen AT, Gomez D, Bell RD, et al. Smooth muscle cell plasticity: fact or fiction? Circ Res. 2013;112:17–22. FREE Full Text
77.↵ Otsuka F, Sakakura K, Yahagi K, Joner M, Virmani R. Has our understanding of calcification in human coronary atherosclerosis progressed? Arterioscler Thromb Vasc Biol. 2014;34:724–736. Abstract/FREE Full Text
78.↵ Stary HC. The development of calcium deposits in atherosclerotic lesions and their persistence after lipid regression. Am J Cardiol. 2001;88:16E–19E. Medline Search Google Scholar
79.↵ Qiao JH, Fishbein MC, Demer LL, Lusis AJ. Genetic determination of cartilaginous metaplasia in mouse aorta. Arterioscler Thromb Vasc Biol. 1995;15:2265–2272. Abstract/FREE Full Text
80.↵ Otsuka F, Finn AV, Virmani R. Do vulnerable and ruptured plaques hide in heavily calcified arteries? Atherosclerosis. 2013;229:34–37. CrossRefMedline Search Google Scholar
81.↵ Glagov S, Weisenberg E, Zarins CK, Stankunavicius R, Kolettis GJ. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med. 1987;316:1371–1375. CrossRefMedline Search Google Scholar
82.↵ Nishioka T, Luo H, Eigler NL, Berglund H, Kim CJ, Siegel RJ. Contribution of inadequate compensatory enlargement to development of human coronary artery stenosis: an in vivo intravascular ultrasound study. J Am Coll Cardiol. 1996;27:1571–1576. CrossRefMedline Search Google Scholar
83.↵ Varnava AM, Mills PG, Davies MJ. Relationship between coronary artery remodeling and plaque vulnerability. Circulation. 2002;105:939–943. Abstract/FREE Full Text
84.↵ Burke AP, Kolodgie FD, Farb A, Weber D, Virmani R. Morphological predictors of arterial remodeling in coronary atherosclerosis. Circulation. 2002;105:297–303. Abstract/FREE Full Text
85.↵ Davies MJ. The pathophysiology of acute coronary syndromes. Heart. 2000;83:361–366. FREE Full Text
86.↵ Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation. 1995;92:657–671. FREE Full Text
87.↵ Schaar JA, Muller JE, Falk E, Virmani R, Fuster V, Serruys PW, Colombo A, Stefanadis C, Ward Casscells S, Moreno PR, Maseri A, van der Steen AF. Terminology for high-risk and vulnerable coronary artery plaques. Report of a meeting on the vulnerable plaque, June 17 and 18, 2003, Santorini, Greece. Eur Heart J. 2004;25:1077–1082. Abstract/FREE Full Text
88.↵ Falk E, Nakano M, Bentzon JF, Finn AV, Virmani R. Update on acute coronary syndromes: the pathologists’ view. Eur Heart J. 2013;34:719–728. Abstract/FREE Full Text
89.↵ Kubo T, Imanishi T, Takarada S, Kuroi A, Ueno S, Yamano T, Tanimoto T, Matsuo Y, Masho T, Kitabata H, Tsuda K, Tomobuchi Y, Akasaka T. Assessment of culprit lesion morphology in acute myocardial infarction: ability of optical coherence tomography compared with intravascular ultrasound and coronary angioscopy. J Am Coll Cardiol. 2007;50:933–939. CrossRefMedline Search Google Scholar
90.↵ Burke AP, Farb A, Malcom GT, Liang Y, Smialek J, Virmani R. Effect of risk factors on the mechanism of acute thrombosis and sudden coronary death in women. Circulation. 1998;97:2110–2116. Abstract/FREE Full Text
91.↵ Burke AP, Farb A, Malcom GT, Liang YH, Smialek J, Virmani R. Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N Engl J Med. 1997;336:1276–1282. CrossRefMedline Search Google Scholar
92.↵ Jia H, Abtahian F, Aguirre AD, et al. In vivo diagnosis of plaque erosion and calcified nodule in patients with acute coronary syndrome by intravascular optical coherence tomography. J Am Coll Cardiol. 2013;62:1748–1758. CrossRef Search Google Scholar
93.↵ Kolodgie FD, Burke AP, Farb A, Gold HK, Yuan J, Narula J, Finn AV, Virmani R. The thin-cap fibroatheroma: a type of vulnerable plaque: the major precursor lesion to acute coronary syndromes. Curr Opin Cardiol. 2001;16:285–292. CrossRefMedline Search Google Scholar
94.↵ van der Wal AC, Becker AE, van der Loos CM, Das PK. Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation. 1994;89:36–44. Abstract/FREE Full Text
95.↵ Gough PJ, Gomez IG, Wille PT, Raines EW. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J Clin Invest. 2006;116:59–69. CrossRefMedline Search Google Scholar
96.↵ Mittleman MA, Mostofsky E. Physical, psychological and chemical triggers of acute cardiovascular events: preventive strategies. Circulation. 2011;124:346–354. FREE Full Text
97.↵ Muller JE, Stone PH, Turi ZG, Rutherford JD, Czeisler CA, Parker C, Poole WK, Passamani E, Roberts R, Robertson T. Circadian variation in the frequency of onset of acute myocardial infarction. N Engl J Med. 1985;313:1315–1322. CrossRefMedline Search Google Scholar
98.↵ Martin JF, Kristensen SD, Mathur A, Grove EL, Choudry FA. The causal role of megakaryocyte–platelet hyperactivity in acute coronary syndromes. Nat Rev Cardiol. 2012;9:658–670. CrossRefMedline Search Google Scholar
99.↵ Kloner RA, Leor J, Poole WK, Perritt R. Population-based analysis of the effect of the Northridge Earthquake on cardiac death in Los Angeles County, California. J Am Coll Cardiol. 1997;30:1174–1180. CrossRefMedline Search Google Scholar
100.↵ Kramer MC, Rittersma SZ, de Winter RJ, Ladich ER, Fowler DR, Liang YH, Kutys R, Carter-Monroe N, Kolodgie FD, van der Wal AC, Virmani R. Relationship of thrombus healing to underlying plaque morphology in sudden coronary death. J Am Coll Cardiol. 2010;55:122–132. CrossRefMedline Search Google Scholar
101.↵ Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol. 2006;47:C13–C18. CrossRefMedline Search Google Scholar
102.↵ Hao H, Gabbiani G, Camenzind E, Bacchetta M, Virmani R, Bochaton-Piallat ML. Phenotypic modulation of intima and media smooth muscle cells in fatal cases of coronary artery lesion. Arterioscler Thromb Vasc Biol. 2006;26:326–332. Abstract/FREE Full Text
103.↵ Davies MJ, Bland JM, Hangartner JR, Angelini A, Thomas AC. Factors influencing the presence or absence of acute coronary artery thrombi in sudden ischaemic death. Eur Heart J. 1989;10:203–208. Abstract/FREE Full Text
104.↵ Mann J, Davies MJ. Mechanisms of progression in native coronary artery disease: role of healed plaque disruption. Heart. 1999;82:265–268. Abstract/FREE Full Text
105.↵ Burke AP, Kolodgie FD, Farb A, Weber DK, Malcom GT, Smialek J, Virmani R. Healed plaque ruptures and sudden coronary death: evidence that subclinical rupture has a role in plaque progression. Circulation. 2001;103:934–940. Abstract/FREE Full Text
106.↵ Barua RS, Ambrose JA. Mechanisms of coronary thrombosis in cigarette smoke exposure. Arterioscler Thromb Vasc Biol. 2013;33:1460–1467. Abstract/FREE Full Text
107.↵ Fernández-Ortiz A, Badimon JJ, Falk E, Fuster V, Meyer B, Mailhac A, Weng D, Shah PK, Badimon L. Characterization of the relative thrombogenicity of atherosclerotic plaque components: implications for consequences of plaque rupture. J Am Coll Cardiol. 1994;23:1562–1569. CrossRefMedline Search Google Scholar
108.↵ Rautou PE, Vion AC, Amabile N, Chironi G, Simon A, Tedgui A, Boulanger CM. Microparticles, vascular function, and atherothrombosis. Circ Res. 2011;109:593–606. Abstract/FREE Full Text
109.↵ Falk E. Unstable angina with fatal outcome: dynamic coronary thrombosis leading to infarction and/or sudden death. Autopsy evidence of recurrent mural thrombosis with peripheral embolization culminating in total vascular occlusion. Circulation. 1985;71:699–708. Abstract/FREE Full Text
110.↵ Rittersma SZ, van der Wal AC, Koch KT, Piek JJ, Henriques JP, Mulder KJ, Ploegmakers JP, Meesterman M, de Winter RJ. Plaque instability frequently occurs days or weeks before occlusive coronary thrombosis: a pathological thrombectomy study in primary percutaneous coronary intervention. Circulation. 2005;111:1160–1165. Abstract/FREE Full Text
111.↵ Wakefield TW, Strieter RM, Wilke CA, Kadell AM, Wrobleski SK, Burdick MD, Schmidt R, Kunkel SL, Greenfield LJ. Venous thrombosis-associated inflammation and attenuation with neutralizing antibodies to cytokines and adhesion molecules. Arterioscler Thromb Vasc Biol. 1995;15:258–268. Abstract/FREE Full Text
112.↵ Falk E, Thuesen L. Pathology of coronary microembolisation and no reflow. Heart. 2003;89:983–985. FREE Full Text
113.↵ Schwartz RS, Burke A, Farb A, Kaye D, Lesser JR, Henry TD, Virmani R. Microemboli and microvascular obstruction in acute coronary thrombosis and sudden coronary death: relation to epicardial plaque histopathology. J Am Coll Cardiol. 2009;54:2167–2173. CrossRefMedline Search Google Scholar
114.↵ Yee KO, Schwartz SM. Why atherosclerotic vessels narrow: the fibrin hypothesis. Thromb Haemost. 1999;82:762–771. Medline Search Google Scholar
115.↵ Bruschke AV, Kramer JR Jr., Bal ET, Haque IU, Detrano RC, Goormastic M. The dynamics of progression of coronary atherosclerosis studied in 168 medically treated patients who underwent coronary arteriography three times. Am Heart J. 1989;117:296–305. CrossRefMedline Search Google Scholar
116.↵ Dalager S, Falk E, Kristensen IB, Paaske WP. Plaque in superficial femoral arteries indicates generalized atherosclerosis and vulnerability to coronary death: an autopsy study. J Vasc Surg. 2008;47:296–302. CrossRefMedline Search Google Scholar
117.↵ Otsuka F, Fuster V, Narula J, Virmani R. Omnipresent atherosclerotic disease: time to depart from analysis of individual vascular beds. Mt Sinai J Med. 2012;79:641–653. CrossRef Search Google Scholar
118.↵ Sillesen H, Muntendam P, Adourian A, Entrekin R, Garcia M, Falk E, Fuster V. Carotid plaque burden as a measure of subclinical atherosclerosis: comparison with other tests for subclinical arterial disease in the High Risk Plaque BioImage study. J Am Coll Cardiol Cardiovasc Imaging. 2012;5:681–689. CrossRef Search Google Scholar
119.↵ National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002;106:3143–3421. FREE Full Text
120.↵ Gonçalves I, Stenström K, Skog G, Mattsson S, Nitulescu M, Nilsson J. Short communication: dating components of human atherosclerotic plaques. Circ Res. 2010;106:1174–1177. Abstract/FREE Full Text
121.↵ Nicholls SJ, Ballantyne CM, Barter PJ, Chapman MJ, Erbel RM, Libby P, Raichlen JS, Uno K, Borgman M, Wolski K, Nissen SE. Effect of two intensive statin regimens on progression of coronary disease. N Engl J Med. 2011;365:2078–2087. CrossRefMedline Search Google Scholar
122.↵ Kini AS, Baber U, Kovacic JC, Limaye A, Ali ZA, Sweeny J, Maehara A, Mehran R, Dangas G, Mintz GS, Fuster V, Narula J, Sharma SK, Moreno PR. Changes in plaque lipid content after short-term intensive versus standard statin therapy: the YELLOW trial (reduction in yellow plaque by aggressive lipid-lowering therapy). J Am Coll Cardiol. 2013;62:21–29. CrossRefMedline Search Google Scholar
123.↵ Björkegren JL, Hägg S, Talukdar HA, Foroughi Asl H, Jain RK, Cedergren C, Shang MM, Rossignoli A, Takolander R, Melander O, Hamsten A, Michoel T, Skogsberg J. Plasma cholesterol-induced lesion networks activated before regression of early, mature, and advanced atherosclerosis. PLoS Genet. 2014;10:e1004201. CrossRef Search Google Scholar
124.↵ Falk E. Pathogenesis of atherosclerosis. J Am Coll Cardiol. 2006;47:C7–C12. CrossRefMedline Search Google Scholar
125.↵ Naghavi M, Libby P, Falk E, et al. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: Part I. Circulation. 2003;108:1664–1672. Abstract/FREE Full Text
126.↵ Wang JC, Normand SL, Mauri L, Kuntz RE. Coronary artery spatial distribution of acute myocardial infarction occlusions. Circulation. 2004;110:278–284. Abstract/FREE Full Text
127.↵ Cheruvu PK, Finn AV, Gardner C, Caplan J, Goldstein J, Stone GW, Virmani R, Muller JE. Frequency and distribution of thin-cap fibroatheroma and ruptured plaques in human coronary arteries: a pathologic study. J Am Coll Cardiol. 2007;50:940–949. CrossRefMedline Search Google Scholar
128.↵ Mauriello A, Sangiorgi G, Fratoni S, Palmieri G, Bonanno E, Anemona L, Schwartz RS, Spagnoli LG. Diffuse and active inflammation occurs in both vulnerable and stable plaques of the entire coronary tree: a histopathologic study of patients dying of acute myocardial infarction. J Am Coll Cardiol. 2005;45:1585–1593. CrossRefMedline Search Google Scholar
129.↵ Stone GW, Maehara A, Lansky AJ, de Bruyne B, Cristea E, Mintz GS, Mehran R, McPherson J, Farhat N, Marso SP, Parise H, Templin B, White R, Zhang Z, Serruys PW; PROSPECT Investigators. A prospective natural-history study of coronary atherosclerosis. N Engl J Med. 2011;364:226–235. CrossRefMedline Search Google Scholar
130.↵ Kubo T, Maehara A, Mintz GS, et al. The dynamic nature of coronary artery lesion morphology assessed by serial virtual histology intravascular ultrasound tissue characterization. J Am Coll Cardiol. 2010;55:1590–1597. CrossRefMedline Search Google Scholar
131.↵ Thim T, Hagensen MK, Wallace-Bradley D, Granada JF, Kaluza GL, Drouet L, Paaske WP, Bøtker HE, Falk E. Unreliable assessment of necrotic core by virtual histology intravascular ultrasound in porcine coronary artery disease. Circ Cardiovasc Imaging. 2010;3:384–391. Abstract/FREE Full Text
132.↵ Falk E, Wilensky RL. Prediction of coronary events by intravascular imaging. J Am Coll Cardiol Cardiovasc Imaging. 2012;5:S38–S41. CrossRef Search Google Scholar
133.↵ Dohi T, Mintz GS, McPherson JA, de Bruyne B, Farhat NZ, Lansky AJ, Mehran R, Weisz G, Xu K, Stone GW, Maehara A. Non-fibroatheroma lesion phenotype and long-term clinical outcomes. J Am Coll Cardiol Cardiovasc Imaging. 2013;6:908–916. CrossRef Search Google Scholar
134.↵ Gertz SD, Roberts WC. Hemodynamic shear force in rupture of coronary arterial atherosclerotic plaques. Am J Cardiol. 1990;66:1368–1372. CrossRefMedline Search Google Scholar
135.↵ Fishbein MC, Siegel RJ. How big are coronary atherosclerotic plaques that rupture? Circulation. 1996;94:2662–2666. FREE Full Text
136.↵ Bezerra HG, Higuchi ML, Gutierrez PS, Palomino SA, Silvestre JM, Libby P, Ramires JA. Atheromas that cause fatal thrombosis are usually large and frequently accompanied by vessel enlargement. Cardiovasc Pathol. 2001;10:189–196. CrossRefMedline Search Google Scholar
137.↵ Puri R, Nicholls SJ, Ellis SG, Tuzcu EM, Kapadia SR. High-risk coronary atheroma—the interplay between ischemia, plaque burden and disease progression. J Am Coll Cardiol. 2014;63:1134–1140. CrossRef Search Google Scholar
138.↵ Niccoli G, Stefanini GG, Capodanno D, Crea F, Ambrose JA, Berg R. Are the culprit lesions severely stenotic? J Am Coll Cardiol Cardiovasc Imaging. 2013;6:1108–1114. Search Google Scholar
139.↵ Narula J, Nakano M, Virmani R, Kolodgie FD, Petersen R, Newcomb R, Malik S, Fuster V, Finn AV. Histopathologic characteristics of atherosclerotic coronary disease and implications of the findings for the invasive and noninvasive detection of vulnerable plaques. J Am Coll Cardiol. 2013;61:1041–1051. CrossRefMedline Search Google Scholar
140.↵ Virmani R, Kolodgie FD, Burke AP, Finn AV, Gold HK, Tulenko TN, Wrenn SP, Narula J. Atherosclerotic plaque progression and vulnerability to rupture: angiogenesis as a source of intraplaque hemorrhage. Arterioscler Thromb Vasc Biol. 2005;25:2054–2061. Abstract/FREE Full Text
141.↵ Kohchi K, Takebayashi S, Hiroki T, Nobuyoshi M. Significance of adventitial inflammation of the coronary artery in patients with unstable angina: results at autopsy. Circulation. 1985;71:709–716. Abstract/FREE Full Text
142.↵ Ehara S, Kobayashi Y, Yoshiyama M, Shimada K, Shimada Y, Fukuda D, Nakamura Y, Yamashita H, Yamagishi H, Takeuchi K, Naruko T, Haze K, Becker AE, Yoshikawa J, Ueda M. Spotty calcification typifies the culprit plaque in patients with acute myocardial infarction: an intravascular ultrasound study. Circulation. 2004;110:3424–3429. Abstract/FREE Full Text
143.↵ Sachdeva A, Cannon CP, Deedwania PC, Labresh KA, Smith SC Jr., Dai D, Hernandez A, Fonarow GC. Lipid levels in patients hospitalized with coronary artery disease: an analysis of 136,905 hospitalizations in Get With The Guidelines. Am Heart J. 2009;157:111.e2–117.e2. Search Google Scholar
144.↵ Sillesen H, Falk E. Why not screen for subclinical atherosclerosis? Lancet. 2011;378:645–646. CrossRefMedline Search Google Scholar
145.↵ Grundy SM. Treatment targets in the management of dyslipidemias: which targets in whom? Curr Cardiol Rep. 2012;14:692–700. CrossRefMedline Search Google Scholar
2.↵ Murray CJ, Lopez AD. Measuring the global burden of disease. N Engl J Med. 2013;369:448–457. CrossRefMedline Search Google Scholar
3.↵ Laslett LJ, Alagona P Jr., Clark BA III., Drozda JP Jr., Saldivar F, Wilson SR, Poe C, Hart M. The worldwide environment of cardiovascular disease: prevalence, diagnosis, therapy, and policy issues: a report from the American College of Cardiology. J Am Coll Cardiol. 2012;60:S1–S49. CrossRefMedline Search Google Scholar
4.↵ Fuster V, Mearns BM. The CVD paradox: mortality vs prevalence. Nat Rev Cardiol. 2009;6:669. CrossRefMedline Search Google Scholar
5.↵ Nabel EG, Braunwald E. A tale of coronary artery disease and myocardial infarction. N Engl J Med. 2012;366:54–63. CrossRefMedline Search Google Scholar
6.↵ Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A, Lanas F, McQueen M, Budaj A, Pais P, Varigos J, Lisheng L; INTERHEART Study Investigators. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet. 2004;364:937–952. CrossRefMedline Search Google Scholar
7.↵ Lim SS, Vos T, Flaxman AD, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2224–2260. CrossRefMedline Search Google Scholar
8.↵ Steinberg D, Glass CK, Witztum JL. Evidence mandating earlier and more aggressive treatment of hypercholesterolemia. Circulation. 2008;118:672–677. FREE Full Text
9.↵ Ference BA, Yoo W, Alesh I, Mahajan N, Mirowska KK, Mewada A, Kahn J, Afonso L, Williams KA, Flack JM. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease. J Am Coll Cardiol. 2012;60:2631–2639. CrossRefMedline Search Google Scholar
10.↵ Bøttcher M, Falk E. Pathology of the coronary arteries in smokers and non-smokers. J Cardiovasc Risk. 1999;6:299–302. Medline Search Google Scholar
11.↵ Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics–2014 update: a report from the American Heart Association. Circulation. 2014;129:e28–e292. FREE Full Text
12.↵ Fowkes FG, Rudan D, Rudan I, Aboyans V, Denenberg JO, McDermott MM, Norman PE, Sampson UK, Williams LJ, Mensah GA, Criqui MH. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: a systematic review and analysis. Lancet. 2013;382:1329–1340. CrossRefMedline Search Google Scholar
13.↵ Willey J, Gonzalez-Castellon M. Cholesterol level and stroke: a complex relationship. JAMA Intern Med. 2013;173:1765–1766. CrossRef Search Google Scholar
14.↵ Bentzon JF, Falk E. Atherosclerotic lesions in mouse and man: is it the same disease? Curr Opin Lipidol. 2010;21:434–440. CrossRefMedline Search Google Scholar
15.↵ Stary HC, Blankenhorn DH, Chandler AB, Glagov S, Insull W Jr., Richardson M, Rosenfeld ME, Schaffer SA, Schwartz CJ, Wagner WD. A definition of the intima of human arteries and of its atherosclerosis-prone regions. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1992;85:391–405. FREE Full Text
16.↵ Stary HC, Chandler AB, Glagov S, Guyton JR, Insull W Jr., Rosenfeld ME, Schaffer SA, Schwartz CJ, Wagner WD, Wissler RW. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler Thromb. 1994;14:840–856. Abstract/FREE Full Text
17.↵ Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W Jr., Rosenfeld ME, Schwartz CJ, Wagner WD, Wissler RW. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1995;92:1355–1374. Abstract/FREE Full Text
18.↵ Stary HC. Natural history and histological classification of atherosclerotic lesions: an update. Arterioscler Thromb Vasc Biol. 2000;20:1177–1178. FREE Full Text
19.↵ Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20:1262–1275. FREE Full Text
20.↵ Wentzel JJ, Chatzizisis YS, Gijsen FJ, Giannoglou GD, Feldman CL, Stone PH. Endothelial shear stress in the evolution of coronary atherosclerotic plaque and vascular remodelling: current understanding and remaining questions. Cardiovasc Res. 2012;96:234–243. Abstract/FREE Full Text
21.↵ Millonig G, Niederegger H, Rabl W, Hochleitner BW, Hoefer D, Romani N, Wick G. Network of vascular-associated dendritic cells in intima of healthy young individuals. Arterioscler Thromb Vasc Biol. 2001;21:503–508. Abstract/FREE Full Text
22.↵ Schwartz SM, deBlois D, O’Brien ER. The intima. Soil for atherosclerosis and restenosis. Circ Res. 1995;77:445–465. FREE Full Text
23.↵ Velican D, Velican C. Atherosclerotic involvement of the coronary arteries of adolescents and young adults. Atherosclerosis. 1980;36:449–460. CrossRefMedline Search Google Scholar
24.↵ Roberts WC. Coronary atherosclerosis: is the process focal or diffuse among patients with symptomatic or fatal myocardial ischemia? Am J Cardiol. 1998;82:41T–44T. Medline Search Google Scholar
25.↵ Cheng C, Tempel D, van Haperen R, van der Baan A, Grosveld F, Daemen MJ, Krams R, de Crom R. Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation. 2006;113:2744–2753. Abstract/FREE Full Text
26.↵ Steinberg D, Witztum JL. Oxidized low-density lipoprotein and atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:2311–2316. FREE Full Text
27.↵ Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. CrossRefMedline Search Google Scholar
28.↵ Subramanian M, Tabas I. Dendritic cells in atherosclerosis. Semin Immunopathol. 2014;36:93–102. CrossRef Search Google Scholar
29.↵ Skålén K, Gustafsson M, Rydberg EK, Hultén LM, Wiklund O, Innerarity TL, Borén J. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. 2002;417:750–754. CrossRefMedline Search Google Scholar
30.↵ Tabas I, Williams KJ, Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116:1832–1844. Abstract/FREE Full Text
31.↵ Stamler J, Daviglus ML, Garside DB, Dyer AR, Greenland P, Neaton JD. Relationship of baseline serum cholesterol levels in 3 large cohorts of younger men to long-term coronary, cardiovascular, and all-cause mortality and to longevity. JAMA. 2000;284:311–318. CrossRefMedline Search Google Scholar
32.↵ Hartiala O, Magnussen CG, Kajander S, et al. Adolescence risk factors are predictive of coronary artery calcification at middle age: the cardiovascular risk in young Finns study. J Am Coll Cardiol. 2012;60:1364–1370. CrossRefMedline Search Google Scholar
33.↵ Leitinger N, Schulman IG. Phenotypic polarization of macrophages in atherosclerosis. Arterioscler Thromb Vasc Biol. 2013;33:1120–1126. Abstract/FREE Full Text
34.↵ Bouhlel MA, Derudas B, Rigamonti E, Dièvart R, Brozek J, Haulon S, Zawadzki C, Jude B, Torpier G, Marx N, Staels B, Chinetti-Gbaguidi G. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–143. CrossRefMedline Search Google Scholar
35.↵ Stöger JL, Gijbels MJ, van der Velden S, Manca M, van der Loos CM, Biessen EA, Daemen MJ, Lutgens E, de Winther MP. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis. 2012;225:461–468. CrossRefMedline Search Google Scholar
36.↵ Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10:36–46. CrossRefMedline Search Google Scholar
37.↵ Hansson GK, Jonasson L. The discovery of cellular immunity in the atherosclerotic plaque. Arterioscler Thromb Vasc Biol. 2009;29:1714–1717. Abstract/FREE Full Text
38.↵ Hansson GK, Nilsson J. Vaccination against atherosclerosis? Induction of atheroprotective immunity. Semin Immunopathol. 2009;31:95–101. CrossRefMedline Search Google Scholar
39.↵ Steinberg D. The LDL modification hypothesis of atherogenesis: an update. J Lipid Res. 2009;50(suppl):S376–S381. Abstract/FREE Full Text
40.↵ Kunjathoor VV, Febbraio M, Podrez EA, Moore KJ, Andersson L, Koehn S, Rhee JS, Silverstein R, Hoff HF, Freeman MW. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem. 2002;277:49982–49988. Abstract/FREE Full Text
41.↵ Haka AS, Grosheva I, Chiang E, Buxbaum AR, Baird BA, Pierini LM, Maxfield FR. Macrophages create an acidic extracellular hydrolytic compartment to digest aggregated lipoproteins. Mol Biol Cell. 2009;20:4932–4940. Abstract/FREE Full Text
42.↵ Witztum JL. You are right too! J Clin Invest. 2005;115:2072–2075. CrossRefMedline Search Google Scholar
43.↵ Katsuda S, Boyd HC, Fligner C, Ross R, Gown AM. Human atherosclerosis. III. Immunocytochemical analysis of the cell composition of lesions of young adults. Am J Pathol. 1992;140:907–914. Medline Search Google Scholar
44.↵ Napoli C, D’Armiento FP, Mancini FP, Postiglione A, Witztum JL, Palumbo G, Palinski W. Fatty streak formation occurs in human fetal aortas and is greatly enhanced by maternal hypercholesterolemia. Intimal accumulation of low density lipoprotein and its oxidation precede monocyte recruitment into early atherosclerotic lesions. J Clin Invest. 1997;100:2680–2690. CrossRefMedline Search Google Scholar
45.↵ Stary HC. Lipid and macrophage accumulations in arteries of children and the development of atherosclerosis. Am J Clin Nutr. 2000;72:1297S–1306S. Abstract/FREE Full Text
46.↵ Kolodgie FD, Burke AP, Nakazawa G, Virmani R. Is pathologic intimal thickening the key to understanding early plaque progression in human atherosclerotic disease? Arterioscler Thromb Vasc Biol. 2007;27:986–989. FREE Full Text
47.↵ Dalager S, Paaske WP, Kristensen IB, Laurberg JM, Falk E. Artery-related differences in atherosclerosis expression: implications for atherogenesis and dynamics in intima-media thickness. Stroke. 2007;38:2698–2705. Abstract/FREE Full Text
48.↵ Kolodgie FD, Gold HK, Burke AP, Fowler DR, Kruth HS, Weber DK, Farb A, Guerrero LJ, Hayase M, Kutys R, Narula J, Finn AV, Virmani R. Intraplaque hemorrhage and progression of coronary atheroma. N Engl J Med. 2003;349:2316–2325. CrossRefMedline Search Google Scholar
49.↵ Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. CrossRefMedline Search Google Scholar
50.↵ Clarke MC, Bennett MR. Cause or consequence: what does macrophage apoptosis do in atherosclerosis? Arterioscler Thromb Vasc Biol. 2009;29:153–155. FREE Full Text
51.↵ Myoishi M, Hao H, Minamino T, Watanabe K, Nishihira K, Hatakeyama K, Asada Y, Okada K, Ishibashi-Ueda H, Gabbiani G, Bochaton-Piallat ML, Mochizuki N, Kitakaze M. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation. 2007;116:1226–1233. Abstract/FREE Full Text
52.↵ Seimon TA, Nadolski MJ, Liao X, Magallon J, Nguyen M, Feric NT, Koschinsky ML, Harkewicz R, Witztum JL, Tsimikas S, Golenbock D, Moore KJ, Tabas I. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. 2010;12:467–482. CrossRefMedline Search Google Scholar
53.↵ Lutgens E, de Muinck ED, Kitslaar PJ, Tordoir JH, Wellens HJ, Daemen MJ. Biphasic pattern of cell turnover characterizes the progression from fatty streaks to ruptured human atherosclerotic plaques. Cardiovasc Res. 1999;41:473–479. Abstract/FREE Full Text
54.↵ Crisby M, Kallin B, Thyberg J, Zhivotovsky B, Orrenius S, Kostulas V, Nilsson J. Cell death in human atherosclerotic plaques involves both oncosis and apoptosis. Atherosclerosis. 1997;130:17–27. CrossRefMedline Search Google Scholar
55.↵ Schrijvers DM, De Meyer GR, Kockx MM, Herman AG, Martinet W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:1256–1261. Abstract/FREE Full Text
56.↵ Gautier EL, Huby T, Witztum JL, Ouzilleau B, Miller ER, Saint-Charles F, Aucouturier P, Chapman MJ, Lesnik P. Macrophage apoptosis exerts divergent effects on atherogenesis as a function of lesion stage. Circulation. 2009;119:1795–1804. Abstract/FREE Full Text
57.↵ Clarke MC, Figg N, Maguire JJ, Davenport AP, Goddard M, Littlewood TD, Bennett MR. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med. 2006;12:1075–1080. CrossRefMedline Search Google Scholar
58.↵ Guyton JR. Phospholipid hydrolytic enzymes in a ‘cesspool’ of arterial intimal lipoproteins: a mechanism for atherogenic lipid accumulation. Arterioscler Thromb Vasc Biol. 2001;21:884–886. FREE Full Text
59.↵ Haka AS, Grosheva I, Singh RK, Maxfield FR. Plasmin promotes foam cell formation by increasing macrophage catabolism of aggregated low-density lipoprotein. Arterioscler Thromb Vasc Biol. 2013;33:1768–1778. Abstract/FREE Full Text
60.↵ McGill HC Jr., McMahan CA, Malcom GT, Oalmann MC, Strong JP. Effects of serum lipoproteins and smoking on atherosclerosis in young men and women. The PDAY Research Group. Pathobiological Determinants of Atherosclerosis in Youth. Arterioscler Thromb Vasc Biol. 1997;17:95–106. Abstract/FREE Full Text
61.↵ Kumamoto M, Nakashima Y, Sueishi K. Intimal neovascularization in human coronary atherosclerosis: its origin and pathophysiological significance. Hum Pathol. 1995;26:450–456. CrossRefMedline Search Google Scholar
62.↵ Sluimer JC, Kolodgie FD, Bijnens AP, Maxfield K, Pacheco E, Kutys B, Duimel H, Frederik PM, van Hinsbergh VW, Virmani R, Daemen MJ. Thin-walled microvessels in human coronary atherosclerotic plaques show incomplete endothelial junctions relevance of compromised structural integrity for intraplaque microvascular leakage. J Am Coll Cardiol. 2009;53:1517–1527. CrossRefMedline Search Google Scholar
63.↵ Davies MJ, Thomas A. Thrombosis and acute coronary-artery lesions in sudden cardiac ischemic death. N Engl J Med. 1984;310:1137–1140. CrossRefMedline Search Google Scholar
64.↵ Falk E. Plaque rupture with severe pre-existing stenosis precipitating coronary thrombosis. Characteristics of coronary atherosclerotic plaques underlying fatal occlusive thrombi. Br Heart J. 1983;50:127–134. Abstract/FREE Full Text
65.↵ Finn AV, Nakano M, Polavarapu R, Karmali V, Saeed O, Zhao X, Yazdani S, Otsuka F, Davis T, Habib A, Narula J, Kolodgie FD, Virmani R. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J Am Coll Cardiol. 2012;59:166–177. CrossRefMedline Search Google Scholar
66.↵ Purushothaman M, Krishnan P, Purushothaman KR, Baber U, Tarricone A, Perez JS, Wiley J, Kini A, Sharma SK, Fuster V, Moreno PR. Genotype-dependent impairment of hemoglobin clearance increases oxidative and inflammatory response in human diabetic atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2769–2775. Abstract/FREE Full Text
67.↵ Cahill LE, Levy AP, Chiuve SE, Jensen MK, Wang H, Shara NM, Blum S, Howard BV, Pai JK, Mukamal KJ, Rexrode KM, Rimm EB. Haptoglobin genotype is a consistent marker of coronary heart disease risk among individuals with elevated glycosylated hemoglobin. J Am Coll Cardiol. 2013;61:728–737. CrossRef Search Google Scholar
68.↵ Kragel AH, Reddy SG, Wittes JT, Roberts WC. Morphometric analysis of the composition of atherosclerotic plaques in the four major epicardial coronary arteries in acute myocardial infarction and in sudden coronary death. Circulation. 1989;80:1747–1756. Abstract/FREE Full Text
69.↵ Stary HC. An Atlas of Atherosclerosis: Progression and Regression. New York: Parthenon Publishing Group; 1999. Search Google Scholar
70.↵ Gomez D, Shankman LS, Nguyen AT, Owens GK. Detection of histone modifications at specific gene loci in single cells in histological sections. Nat Methods. 2013;10:171–177. CrossRefMedline Search Google Scholar
71.↵ Bentzon JF, Weile C, Sondergaard CS, Hindkjaer J, Kassem M, Falk E. Smooth muscle cells in atherosclerosis originate from the local vessel wall and not circulating progenitor cells in ApoE knockout mice. Arterioscler Thromb Vasc Biol. 2006;26:2696–2702. Abstract/FREE Full Text
72.↵ Campbell JH, Campbell GR. Smooth muscle phenotypic modulation—a personal experience. Arterioscler Thromb Vasc Biol. 2012;32:1784–1789. Abstract/FREE Full Text
73.↵ Feil S, Hofmann F, Feil R. SM22alpha modulates vascular smooth muscle cell phenotype during atherogenesis. Circ Res. 2004;94:863–865. Abstract/FREE Full Text
74.↵ Tang Z, Wang A, Yuan F, Yan Z, Liu B, Chu JS, Helms JA, Li S. Differentiation of multipotent vascular stem cells contributes to vascular diseases. Nat Commun. 2012;3:875. CrossRefMedline Search Google Scholar
75.↵ Bentzon JF, Falk E. Circulating smooth muscle progenitor cells in atherosclerosis and plaque rupture: current perspective and methods of analysis. Vascul Pharmacol. 2010;52:11–20. CrossRefMedline Search Google Scholar
76.↵ Nguyen AT, Gomez D, Bell RD, et al. Smooth muscle cell plasticity: fact or fiction? Circ Res. 2013;112:17–22. FREE Full Text
77.↵ Otsuka F, Sakakura K, Yahagi K, Joner M, Virmani R. Has our understanding of calcification in human coronary atherosclerosis progressed? Arterioscler Thromb Vasc Biol. 2014;34:724–736. Abstract/FREE Full Text
78.↵ Stary HC. The development of calcium deposits in atherosclerotic lesions and their persistence after lipid regression. Am J Cardiol. 2001;88:16E–19E. Medline Search Google Scholar
79.↵ Qiao JH, Fishbein MC, Demer LL, Lusis AJ. Genetic determination of cartilaginous metaplasia in mouse aorta. Arterioscler Thromb Vasc Biol. 1995;15:2265–2272. Abstract/FREE Full Text
80.↵ Otsuka F, Finn AV, Virmani R. Do vulnerable and ruptured plaques hide in heavily calcified arteries? Atherosclerosis. 2013;229:34–37. CrossRefMedline Search Google Scholar
81.↵ Glagov S, Weisenberg E, Zarins CK, Stankunavicius R, Kolettis GJ. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med. 1987;316:1371–1375. CrossRefMedline Search Google Scholar
82.↵ Nishioka T, Luo H, Eigler NL, Berglund H, Kim CJ, Siegel RJ. Contribution of inadequate compensatory enlargement to development of human coronary artery stenosis: an in vivo intravascular ultrasound study. J Am Coll Cardiol. 1996;27:1571–1576. CrossRefMedline Search Google Scholar
83.↵ Varnava AM, Mills PG, Davies MJ. Relationship between coronary artery remodeling and plaque vulnerability. Circulation. 2002;105:939–943. Abstract/FREE Full Text
84.↵ Burke AP, Kolodgie FD, Farb A, Weber D, Virmani R. Morphological predictors of arterial remodeling in coronary atherosclerosis. Circulation. 2002;105:297–303. Abstract/FREE Full Text
85.↵ Davies MJ. The pathophysiology of acute coronary syndromes. Heart. 2000;83:361–366. FREE Full Text
86.↵ Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation. 1995;92:657–671. FREE Full Text
87.↵ Schaar JA, Muller JE, Falk E, Virmani R, Fuster V, Serruys PW, Colombo A, Stefanadis C, Ward Casscells S, Moreno PR, Maseri A, van der Steen AF. Terminology for high-risk and vulnerable coronary artery plaques. Report of a meeting on the vulnerable plaque, June 17 and 18, 2003, Santorini, Greece. Eur Heart J. 2004;25:1077–1082. Abstract/FREE Full Text
88.↵ Falk E, Nakano M, Bentzon JF, Finn AV, Virmani R. Update on acute coronary syndromes: the pathologists’ view. Eur Heart J. 2013;34:719–728. Abstract/FREE Full Text
89.↵ Kubo T, Imanishi T, Takarada S, Kuroi A, Ueno S, Yamano T, Tanimoto T, Matsuo Y, Masho T, Kitabata H, Tsuda K, Tomobuchi Y, Akasaka T. Assessment of culprit lesion morphology in acute myocardial infarction: ability of optical coherence tomography compared with intravascular ultrasound and coronary angioscopy. J Am Coll Cardiol. 2007;50:933–939. CrossRefMedline Search Google Scholar
90.↵ Burke AP, Farb A, Malcom GT, Liang Y, Smialek J, Virmani R. Effect of risk factors on the mechanism of acute thrombosis and sudden coronary death in women. Circulation. 1998;97:2110–2116. Abstract/FREE Full Text
91.↵ Burke AP, Farb A, Malcom GT, Liang YH, Smialek J, Virmani R. Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N Engl J Med. 1997;336:1276–1282. CrossRefMedline Search Google Scholar
92.↵ Jia H, Abtahian F, Aguirre AD, et al. In vivo diagnosis of plaque erosion and calcified nodule in patients with acute coronary syndrome by intravascular optical coherence tomography. J Am Coll Cardiol. 2013;62:1748–1758. CrossRef Search Google Scholar
93.↵ Kolodgie FD, Burke AP, Farb A, Gold HK, Yuan J, Narula J, Finn AV, Virmani R. The thin-cap fibroatheroma: a type of vulnerable plaque: the major precursor lesion to acute coronary syndromes. Curr Opin Cardiol. 2001;16:285–292. CrossRefMedline Search Google Scholar
94.↵ van der Wal AC, Becker AE, van der Loos CM, Das PK. Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation. 1994;89:36–44. Abstract/FREE Full Text
95.↵ Gough PJ, Gomez IG, Wille PT, Raines EW. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J Clin Invest. 2006;116:59–69. CrossRefMedline Search Google Scholar
96.↵ Mittleman MA, Mostofsky E. Physical, psychological and chemical triggers of acute cardiovascular events: preventive strategies. Circulation. 2011;124:346–354. FREE Full Text
97.↵ Muller JE, Stone PH, Turi ZG, Rutherford JD, Czeisler CA, Parker C, Poole WK, Passamani E, Roberts R, Robertson T. Circadian variation in the frequency of onset of acute myocardial infarction. N Engl J Med. 1985;313:1315–1322. CrossRefMedline Search Google Scholar
98.↵ Martin JF, Kristensen SD, Mathur A, Grove EL, Choudry FA. The causal role of megakaryocyte–platelet hyperactivity in acute coronary syndromes. Nat Rev Cardiol. 2012;9:658–670. CrossRefMedline Search Google Scholar
99.↵ Kloner RA, Leor J, Poole WK, Perritt R. Population-based analysis of the effect of the Northridge Earthquake on cardiac death in Los Angeles County, California. J Am Coll Cardiol. 1997;30:1174–1180. CrossRefMedline Search Google Scholar
100.↵ Kramer MC, Rittersma SZ, de Winter RJ, Ladich ER, Fowler DR, Liang YH, Kutys R, Carter-Monroe N, Kolodgie FD, van der Wal AC, Virmani R. Relationship of thrombus healing to underlying plaque morphology in sudden coronary death. J Am Coll Cardiol. 2010;55:122–132. CrossRefMedline Search Google Scholar
101.↵ Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol. 2006;47:C13–C18. CrossRefMedline Search Google Scholar
102.↵ Hao H, Gabbiani G, Camenzind E, Bacchetta M, Virmani R, Bochaton-Piallat ML. Phenotypic modulation of intima and media smooth muscle cells in fatal cases of coronary artery lesion. Arterioscler Thromb Vasc Biol. 2006;26:326–332. Abstract/FREE Full Text
103.↵ Davies MJ, Bland JM, Hangartner JR, Angelini A, Thomas AC. Factors influencing the presence or absence of acute coronary artery thrombi in sudden ischaemic death. Eur Heart J. 1989;10:203–208. Abstract/FREE Full Text
104.↵ Mann J, Davies MJ. Mechanisms of progression in native coronary artery disease: role of healed plaque disruption. Heart. 1999;82:265–268. Abstract/FREE Full Text
105.↵ Burke AP, Kolodgie FD, Farb A, Weber DK, Malcom GT, Smialek J, Virmani R. Healed plaque ruptures and sudden coronary death: evidence that subclinical rupture has a role in plaque progression. Circulation. 2001;103:934–940. Abstract/FREE Full Text
106.↵ Barua RS, Ambrose JA. Mechanisms of coronary thrombosis in cigarette smoke exposure. Arterioscler Thromb Vasc Biol. 2013;33:1460–1467. Abstract/FREE Full Text
107.↵ Fernández-Ortiz A, Badimon JJ, Falk E, Fuster V, Meyer B, Mailhac A, Weng D, Shah PK, Badimon L. Characterization of the relative thrombogenicity of atherosclerotic plaque components: implications for consequences of plaque rupture. J Am Coll Cardiol. 1994;23:1562–1569. CrossRefMedline Search Google Scholar
108.↵ Rautou PE, Vion AC, Amabile N, Chironi G, Simon A, Tedgui A, Boulanger CM. Microparticles, vascular function, and atherothrombosis. Circ Res. 2011;109:593–606. Abstract/FREE Full Text
109.↵ Falk E. Unstable angina with fatal outcome: dynamic coronary thrombosis leading to infarction and/or sudden death. Autopsy evidence of recurrent mural thrombosis with peripheral embolization culminating in total vascular occlusion. Circulation. 1985;71:699–708. Abstract/FREE Full Text
110.↵ Rittersma SZ, van der Wal AC, Koch KT, Piek JJ, Henriques JP, Mulder KJ, Ploegmakers JP, Meesterman M, de Winter RJ. Plaque instability frequently occurs days or weeks before occlusive coronary thrombosis: a pathological thrombectomy study in primary percutaneous coronary intervention. Circulation. 2005;111:1160–1165. Abstract/FREE Full Text
111.↵ Wakefield TW, Strieter RM, Wilke CA, Kadell AM, Wrobleski SK, Burdick MD, Schmidt R, Kunkel SL, Greenfield LJ. Venous thrombosis-associated inflammation and attenuation with neutralizing antibodies to cytokines and adhesion molecules. Arterioscler Thromb Vasc Biol. 1995;15:258–268. Abstract/FREE Full Text
112.↵ Falk E, Thuesen L. Pathology of coronary microembolisation and no reflow. Heart. 2003;89:983–985. FREE Full Text
113.↵ Schwartz RS, Burke A, Farb A, Kaye D, Lesser JR, Henry TD, Virmani R. Microemboli and microvascular obstruction in acute coronary thrombosis and sudden coronary death: relation to epicardial plaque histopathology. J Am Coll Cardiol. 2009;54:2167–2173. CrossRefMedline Search Google Scholar
114.↵ Yee KO, Schwartz SM. Why atherosclerotic vessels narrow: the fibrin hypothesis. Thromb Haemost. 1999;82:762–771. Medline Search Google Scholar
115.↵ Bruschke AV, Kramer JR Jr., Bal ET, Haque IU, Detrano RC, Goormastic M. The dynamics of progression of coronary atherosclerosis studied in 168 medically treated patients who underwent coronary arteriography three times. Am Heart J. 1989;117:296–305. CrossRefMedline Search Google Scholar
116.↵ Dalager S, Falk E, Kristensen IB, Paaske WP. Plaque in superficial femoral arteries indicates generalized atherosclerosis and vulnerability to coronary death: an autopsy study. J Vasc Surg. 2008;47:296–302. CrossRefMedline Search Google Scholar
117.↵ Otsuka F, Fuster V, Narula J, Virmani R. Omnipresent atherosclerotic disease: time to depart from analysis of individual vascular beds. Mt Sinai J Med. 2012;79:641–653. CrossRef Search Google Scholar
118.↵ Sillesen H, Muntendam P, Adourian A, Entrekin R, Garcia M, Falk E, Fuster V. Carotid plaque burden as a measure of subclinical atherosclerosis: comparison with other tests for subclinical arterial disease in the High Risk Plaque BioImage study. J Am Coll Cardiol Cardiovasc Imaging. 2012;5:681–689. CrossRef Search Google Scholar
119.↵ National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002;106:3143–3421. FREE Full Text
120.↵ Gonçalves I, Stenström K, Skog G, Mattsson S, Nitulescu M, Nilsson J. Short communication: dating components of human atherosclerotic plaques. Circ Res. 2010;106:1174–1177. Abstract/FREE Full Text
121.↵ Nicholls SJ, Ballantyne CM, Barter PJ, Chapman MJ, Erbel RM, Libby P, Raichlen JS, Uno K, Borgman M, Wolski K, Nissen SE. Effect of two intensive statin regimens on progression of coronary disease. N Engl J Med. 2011;365:2078–2087. CrossRefMedline Search Google Scholar
122.↵ Kini AS, Baber U, Kovacic JC, Limaye A, Ali ZA, Sweeny J, Maehara A, Mehran R, Dangas G, Mintz GS, Fuster V, Narula J, Sharma SK, Moreno PR. Changes in plaque lipid content after short-term intensive versus standard statin therapy: the YELLOW trial (reduction in yellow plaque by aggressive lipid-lowering therapy). J Am Coll Cardiol. 2013;62:21–29. CrossRefMedline Search Google Scholar
123.↵ Björkegren JL, Hägg S, Talukdar HA, Foroughi Asl H, Jain RK, Cedergren C, Shang MM, Rossignoli A, Takolander R, Melander O, Hamsten A, Michoel T, Skogsberg J. Plasma cholesterol-induced lesion networks activated before regression of early, mature, and advanced atherosclerosis. PLoS Genet. 2014;10:e1004201. CrossRef Search Google Scholar
124.↵ Falk E. Pathogenesis of atherosclerosis. J Am Coll Cardiol. 2006;47:C7–C12. CrossRefMedline Search Google Scholar
125.↵ Naghavi M, Libby P, Falk E, et al. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: Part I. Circulation. 2003;108:1664–1672. Abstract/FREE Full Text
126.↵ Wang JC, Normand SL, Mauri L, Kuntz RE. Coronary artery spatial distribution of acute myocardial infarction occlusions. Circulation. 2004;110:278–284. Abstract/FREE Full Text
127.↵ Cheruvu PK, Finn AV, Gardner C, Caplan J, Goldstein J, Stone GW, Virmani R, Muller JE. Frequency and distribution of thin-cap fibroatheroma and ruptured plaques in human coronary arteries: a pathologic study. J Am Coll Cardiol. 2007;50:940–949. CrossRefMedline Search Google Scholar
128.↵ Mauriello A, Sangiorgi G, Fratoni S, Palmieri G, Bonanno E, Anemona L, Schwartz RS, Spagnoli LG. Diffuse and active inflammation occurs in both vulnerable and stable plaques of the entire coronary tree: a histopathologic study of patients dying of acute myocardial infarction. J Am Coll Cardiol. 2005;45:1585–1593. CrossRefMedline Search Google Scholar
129.↵ Stone GW, Maehara A, Lansky AJ, de Bruyne B, Cristea E, Mintz GS, Mehran R, McPherson J, Farhat N, Marso SP, Parise H, Templin B, White R, Zhang Z, Serruys PW; PROSPECT Investigators. A prospective natural-history study of coronary atherosclerosis. N Engl J Med. 2011;364:226–235. CrossRefMedline Search Google Scholar
130.↵ Kubo T, Maehara A, Mintz GS, et al. The dynamic nature of coronary artery lesion morphology assessed by serial virtual histology intravascular ultrasound tissue characterization. J Am Coll Cardiol. 2010;55:1590–1597. CrossRefMedline Search Google Scholar
131.↵ Thim T, Hagensen MK, Wallace-Bradley D, Granada JF, Kaluza GL, Drouet L, Paaske WP, Bøtker HE, Falk E. Unreliable assessment of necrotic core by virtual histology intravascular ultrasound in porcine coronary artery disease. Circ Cardiovasc Imaging. 2010;3:384–391. Abstract/FREE Full Text
132.↵ Falk E, Wilensky RL. Prediction of coronary events by intravascular imaging. J Am Coll Cardiol Cardiovasc Imaging. 2012;5:S38–S41. CrossRef Search Google Scholar
133.↵ Dohi T, Mintz GS, McPherson JA, de Bruyne B, Farhat NZ, Lansky AJ, Mehran R, Weisz G, Xu K, Stone GW, Maehara A. Non-fibroatheroma lesion phenotype and long-term clinical outcomes. J Am Coll Cardiol Cardiovasc Imaging. 2013;6:908–916. CrossRef Search Google Scholar
134.↵ Gertz SD, Roberts WC. Hemodynamic shear force in rupture of coronary arterial atherosclerotic plaques. Am J Cardiol. 1990;66:1368–1372. CrossRefMedline Search Google Scholar
135.↵ Fishbein MC, Siegel RJ. How big are coronary atherosclerotic plaques that rupture? Circulation. 1996;94:2662–2666. FREE Full Text
136.↵ Bezerra HG, Higuchi ML, Gutierrez PS, Palomino SA, Silvestre JM, Libby P, Ramires JA. Atheromas that cause fatal thrombosis are usually large and frequently accompanied by vessel enlargement. Cardiovasc Pathol. 2001;10:189–196. CrossRefMedline Search Google Scholar
137.↵ Puri R, Nicholls SJ, Ellis SG, Tuzcu EM, Kapadia SR. High-risk coronary atheroma—the interplay between ischemia, plaque burden and disease progression. J Am Coll Cardiol. 2014;63:1134–1140. CrossRef Search Google Scholar
138.↵ Niccoli G, Stefanini GG, Capodanno D, Crea F, Ambrose JA, Berg R. Are the culprit lesions severely stenotic? J Am Coll Cardiol Cardiovasc Imaging. 2013;6:1108–1114. Search Google Scholar
139.↵ Narula J, Nakano M, Virmani R, Kolodgie FD, Petersen R, Newcomb R, Malik S, Fuster V, Finn AV. Histopathologic characteristics of atherosclerotic coronary disease and implications of the findings for the invasive and noninvasive detection of vulnerable plaques. J Am Coll Cardiol. 2013;61:1041–1051. CrossRefMedline Search Google Scholar
140.↵ Virmani R, Kolodgie FD, Burke AP, Finn AV, Gold HK, Tulenko TN, Wrenn SP, Narula J. Atherosclerotic plaque progression and vulnerability to rupture: angiogenesis as a source of intraplaque hemorrhage. Arterioscler Thromb Vasc Biol. 2005;25:2054–2061. Abstract/FREE Full Text
141.↵ Kohchi K, Takebayashi S, Hiroki T, Nobuyoshi M. Significance of adventitial inflammation of the coronary artery in patients with unstable angina: results at autopsy. Circulation. 1985;71:709–716. Abstract/FREE Full Text
142.↵ Ehara S, Kobayashi Y, Yoshiyama M, Shimada K, Shimada Y, Fukuda D, Nakamura Y, Yamashita H, Yamagishi H, Takeuchi K, Naruko T, Haze K, Becker AE, Yoshikawa J, Ueda M. Spotty calcification typifies the culprit plaque in patients with acute myocardial infarction: an intravascular ultrasound study. Circulation. 2004;110:3424–3429. Abstract/FREE Full Text
143.↵ Sachdeva A, Cannon CP, Deedwania PC, Labresh KA, Smith SC Jr., Dai D, Hernandez A, Fonarow GC. Lipid levels in patients hospitalized with coronary artery disease: an analysis of 136,905 hospitalizations in Get With The Guidelines. Am Heart J. 2009;157:111.e2–117.e2. Search Google Scholar
144.↵ Sillesen H, Falk E. Why not screen for subclinical atherosclerosis? Lancet. 2011;378:645–646. CrossRefMedline Search Google Scholar
145.↵ Grundy SM. Treatment targets in the management of dyslipidemias: which targets in whom? Curr Cardiol Rep. 2012;14:692–700. CrossRefMedline Search Google Scholar
146.↵ Webber BJ, Seguin PG, Burnett DG, Clark LL, Otto JL. Prevalence of and risk factors for autopsy-determined atherosclerosis among US service members, 2001-2011. JAMA. 2012;308:2577–2583.

No hay comentarios:
Publicar un comentario