
Diabetes subclínica
¿Se está diagnosticando la diabetes demasiado tarde?: Las personas normoglucémicas también tienen posibilidades de sufrir las complicaciones y comorbilidades de la diabetes.

|
La Organización Mundial de la Salud (OMS), define la salud como un estado de completo bienestar físico, mental y también social, no solamente la ausencia de enfermedad. Sin embargo, la manera cómo un individuo o un médico perciban este aparente bienestar depende del contexto social y de los estándares de atención de las enfermedades, que no son conceptos inmutables. Estos valores de referencia se basan sobre la estratificación de los valores hallados frecuentemente en la población y su relación con marcadores del inicio de las enfermedades, que no presuponen necesariamente un “completo” estado de salud.
Los marcadores pueden variar mucho según las distintas sociedades y su predisposición a las enfermedades crónicas. Además, se halló que las comorbilidades vinculadas con una enfermedad se relacionan con la estratificación dentro de los valores normales de los marcadores clínicos, suscitando la inquietud de si una determinada enfermedad crónica podría estar ya en curso mucho antes de su diagnóstico clínico, en una etapa subclínica.
En este artículo se analiza si el concepto de enfermedad subclínica se podría aplicar a la diabetes mellitus tipo 2 (DM2). Además se presentan sugerencias para el diagnóstico más temprano del estado asintomático previo a la enfermedad y para la prevención del riesgo de diabetes, sus complicaciones y comorbilidades.
La DM2 es un conglomerado de enfermedades metabólicas crónicas, en la que la hiperglucemia es un marcador común. También tienen en común el mal funcionamiento del páncreas endócrino, en especial las células beta, que son muy sensibles a diversos factores ambientales, inflamatorios, inmunitarios y genéticos (Schwartz et al. 2016). En la DM tipo 1 (DMT1) con insuficiente producción de insulina debido a la destrucción de las células beta.
La American Diabetes Association (ADA) señala que la DMT2 se debe a la pérdida progresiva de la secreción de insulina en presencia de resistencia a la insulina. En realidad, en la DMT2 la disminución de la masa de células beta y de la producción de insulina es precedida por un estado de hiperinsulinemia, como compensación por el estado subyacente de resistencia a la insulina. (Yalow and Berson 1960, Association 1998, Garber et al. 2016) (WHO 2015).
Varios estudios y organizaciones mostraron el aumento sin precedentes de la prevalencia de diabetes en todo el mundo (NCD-RisC 2016), incluida la DMT1. (Harjutsalo et al. 2008, Patterson et al. 2009, Dabelea et al. 2014, Forga Llenas et al.2015, Lamb et al. 2015). El número estimado de adultos con diabetes aumentó a más de 380 millones (> 8 % de la población adulta mundial) (Menke et al. 2015), con una estimación de más del 37 % con prediabetes y más del 45 % sin diagnosticar (Schmidt et al. 2011, Federation 2015, Mechanick 2015, Menke 2015).
Es necesario el diagnóstico más temprano del aumento del riesgo de diabetes y sus complicaciones y comorbilidades
La prevalencia de DMT2 en personas más jóvenes también aumentó (Holden et al. 2013, Menke et al. 2015), con una nueva clase de pacientes: niños (Lustig et al. 2016), adolescentes y adultos jóvenes (Weiss et al. 2013, Song 2016). Estos grupos de "no adultos" sufren de DMT2 y síndrome metabólico (SMet) diagnosticado por 3 de las 5 mediciones siguientes:
EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG |
Es probable que a corto plazo formarán un nuevo grupo de pacientes adultos jóvenes, con consecuencias de por vida sobre su salud. Es necesario el diagnóstico más temprano del aumento del riesgo de diabetes y sus complicaciones y comorbilidades (Lima 2017), así como intervenciones para revertir estas enfermedades a fin de reducir al mínimo el impacto sobre su salud y sobre el sistema sanitario.
El cuadro siguiente resume los criterios diagnósticos actuales de la ADA (Association 2016) y la OMS (WHO 2016, Colagiuri et al. 2011).
Criterios actuales para el diagnóstico de diabetes y de alteración del metabolismo de la glucosa
GA – glucemia en ayunas (por lo menos 8 hs sin consumo calórico). 2hs POTG– 2hs poscarga de glucosa plasmática tras 75 g de glucosa oral tras por lo menos 8 hs de ayuno. IG – Intolerancia a la glucosa. AGA – Alteración de la glucosa en ayunas. * HbA1c – aunque se especifica un intervalo de referencia en consonancia con los criterios de la ADA, el SBD no adopta la HbA1c como criterio diagnóstico de diabetes. La glucemia se puede convertir de mg/dl a mM dividiendo por 18. EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG |

Desde 2001 se emplea un nuevo conjunto de criterios para el diagnóstico de diabetes. Según estos la diabetes manifiesta es precedida por dos etapas de no diabetes:
i) pre-diabetes: estado alterado (alteración de la glucosa en ayunas o intolerancia a la glucosa) donde estos marcadores están alterados hacia el nivel de la diabetes y
ii) no-diabéticos: dentro del intervalo de referencia normal (incluida la normoglucemia glucemia en ayunas < 100 mg/dl, prueba oral de tolerancia a la glucosa (POTG) <140 mg/ml y HbA1c < 5.7 %).
EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG
|
A pesar del amplio empleo de estos marcadores clínicos, se cuestiona si constituyen un grupo de análisis satisfactorios para el diagnóstico de diabetes y prediabetes o el pronóstico temprano de diabetes a futuro (Zhang et al. 2005, Toscano et al. 2015).
A fin de evaluar el aumento del riesgo de diabetes, la ADA recomienda estudiar a todas las personas > 45 años o con sobrepeso u obesidad (IMC > 25 kg/m2) u otros factores de riesgo, como inactividad física, un familiar de primer grado con diabetes, hipertensión (≥ 140/90 mm Hg o en tratamiento por hipertensión) o antecedentes de enfermedad cardiovascular (ECV), C-HDL < 35 mg/dl o triglicéridos > 250 mg/ dl (2,82 mmol/l o en tratamiento por dislipidemia), entre otras (Association 2016).
Sin embargo, cuando se los diagnostica, los pacientes con DMT2 ya tienen disminuida alrededor del 50 % la función de las células beta del páncreas (DeWitt and Hirsch, 2003), así como signos de la prevalencia de marcadores de complicaciones de la diabetes, como microalbuminuria debida a daño renal (UKPDS 1998a, Stratton et al. 2000, Bash et al. 2008) y retinopatía (Nagi et al. 1997, Kohner et al. 1998, Looker et al. 2012).
Los criterios clínicos “glucocéntricos” (Yudkin and Montori 2014) quizás constituyan un diagnóstico tardío
En el United Kingdom Prospective Diabetes Study (UKPDS), todos los resultados microvasculares y macrovasculares se relacionaron con la HbA1c como un continuo positivo, lineal (Stratton et al. 2000). La gravedad creciente de la retinopatía se asoció con valores menores de insulinemia (Kohner et al. 1998), lo que sugiere el diagnóstico tardío de la diabetes y se relaciona con una gran pérdida de la función de las células beta en el momento del diagnóstico (DeWitt and Hirsch 2003).
Estos datos sugieren que las complicaciones de la diabetes se pueden observar desde que se la diagnostica y por lo tanto los criterios clínicos “glucocéntricos” (Yudkin and Montori 2014) quizás constituyan un diagnóstico tardío. Indican así la necesidad de detección más precoz de la resistencia a la insulina y de la alteración de la función pancreática.
▶ Con un margen normoglucémico
Varios estudios prospectivos indicaron que la incidencia de diabetes coincidía progresivamente con la glucemia en ayunas dentro del margen normoglucémico y aumentaba para los pacientes con glucemia en ayunas mayor de ese nivel (Shaw et al. 2000). (Tirosh et al. 2005 (Kato et al. 2009).
En un gran estudio de Kaiser Permanente Northwest, EEUU, se siguió durante 10 años a más de 46000 personas > 40 años. El riesgo de sufrir diabetes se relacionó con la glucemia en ayunas dentro de los valores normales, pero fue mayor para aquellos en los cuartilos superiores. Los pacientes que sufrieron diabetes también tenían mayor IMC y valores más altos de triglicéridos, con C-HDL ligeramente disminuido, características todas de dislipidemia y padecían hipertensión y enfermedad cardiovascular. Estos datos indican no solo la importancia de evaluar los indicadores glucémicos, sino también la importancia de las tendencias en los marcadores del SMet y de resistencia a la insulina.
Hasta que se la diagnostica, la diabetes puede pasar desapercibida durante más de una década con los criterios diagnósticos actuales. Otros varios estudios muestran que el diagnóstico de diabetes está precedido por más de una década de progresión asintomática de la disfunción metabólica, incluida la disminución de la sensibilidad a la insulina y de la función de las células beta.
En conjunto, todos estos estudios evidencian que el riesgo de diabetes a futuro aumenta significativamente en función de los marcadores glucémicos y de la función de las células beta, como un continuo que comienza dentro del grupo de personas normoglucémicas.
▶ Riesgos de complicaciones de la diabetes y comorbilidades dentro del intervalo de referencia normal
◈ Complicaciones de la diabetes en pacientes diabéticos
Varios estudios mostraron el riesgo de sufrir complicaciones de la diabetes en pacientes diabéticos sujetos al control glucémico estricto con protocolos de tratamiento farmacológico intensivo.
El Diabetes Control and Complication Trial (DCCT) efectuado en 1441 personas con DMT1 con una media de seguimiento de 6,5 años mostró que en las condiciones arriba mencionadas disminuyeron las complicaciones a largo plazo, como la retinopatía, la nefropatía y la neuropatía (DCCT 1993). Estos beneficios tuvieron efectos prolongados sostenidos, observados en el DCCT-EDIC 2000) así como en otros estudios.
El UK Prospective Diabetes Study (UKPDS) con pacientes con DMT2 en seguimiento durante 10 años mostró que el control estricto de la glucemia disminuyó el riesgo de ECV, mortalidad por todas las causas, enfermedades microvasculares y macrovasculares.
◈ Complicaciones de la diabetes en individuos no diabéticos
Los riesgos de complicaciones de la diabetes también se observaron como resultado de disfunción metabólica entre personas normoglucémicas.
♦ Retinopatía
La tendencia al aumento de la prevalencia de retinopatía se observó como una variable dependiente de la HbA1c dentro del límite normal de referencia en algunos estudios y en el National Health and Nutrition Examination Survey (NHANES) (WHO 2015, Colagiuri et al. 2011).
♦ Enfermedad cardiovascular
En el estudio San Antonio Heart Study, se efectuó el seguimiento durante ocho años de más de 2560 personas no diabéticas al inicio. El riesgo de ECV se evaluó y se halló que el HOMA-IR y la insulinemia en ayunas eran un factor de riesgo independiente con la glucemia en ayunas y la prueba oral de tolerancia a la glucosa (POTG) dentro de los límites normales.
El diagnóstico de DMT2 se consideró un factor de riesgo independiente para ECV (Valenti et al. 2016). El estudio prospectivo DECODE con 22 cohortes europeas y más de 29700 personas con una mediana de seguimiento de 11 años informó riesgo en aumento para la mortalidad por ECV y no ECV para personas con valores de la POTG dentro del intervalo de normoglucemia. Resultados similares se observaron en numerosos estudios de distintos países
♦ Cáncer
En un estudio prospectivo japonés que siguió a más de 29.000 personas sin cáncer al inicio, se halló aumento del riesgo de cáncer en los que tenían valores de HbA1c más altos, dentro de los intervalos no diabéticos y diabéticos (Goto et al. 2016).
En un estudio prospectivo italiano con más de 10000 personas seguidas durante 5 años, se halló que la glucemia en ayunas dentro del intervalo de referencia de los no diabéticos se asoció con aumento del riesgo de cáncer de mama.
♦ Demencia
Durante una mediana de seguimiento de 6,8 años de más de 2000 pacientes, el estudio Adult Changes in Thought (ACT) halló un riesgo progresivo creciente de demencia en función de la glucemia al inicio en grupos sin diabetes (Crane et al. 2013), sugiriendo resistencia a la insulina y aumento de complicaciones microvasculares en el sistema nervioso central como causas subyacentes.
En otro estudio, con adultos > 65 años y sin signos de demencia al inicio, las probabilidades de enfermedad de Alzheimer y demencia aumentaron a más del 100 % en las personas con hiperinsulinemia (Luchsinger et al. 2004).
♦ Enfermedades renales
En el estudio Atherosclerosis Risk in Communities, con 1800 participantes diabéticos, se halló una asociación positiva entre la nefropatía crónica - en ausencia de albuminuria y retinopatía – y una amplia gama de valores de HbA1c , incluidos los valores normales de referencia (<5,7 %)
En conjunto, todos estos datos ilustran que los riesgos de complicaciones relacionadas con la diabetes y comorbilidades se relacionan con marcadores del metabolismo de la glucosa de manera dosis-dependiente dentro de los intervalos de referencia normales para los no diabéticos. Por consiguiente, estos datos indican que existen manifestaciones iniciales de complicaciones relacionadas con la diabetes subclínica no detectadas por el enfoque diagnóstico actual.
▶ Hiperhormonemia y resistencia a la insulina como elementos faltantes
♦ Hiperhormonemia pancreática como patognomónica de la diabetes
Tras el diagnóstico, la progresión de la DMT2 se caracteriza por el deterioro de la función de las células beta pancreáticas (Butler et al. 2003, DeWitt and Hirsch 2003, Association 2016). Este deterioro, así como el de la secreción de insulina con la progresión de la prediabetes y la diabetes manifiesta, es precedido por un estado de hiperhormonemia en el que la secreción hormonal (insulina y amilina) de las células beta intenta compensar la carga de glucosa y el grado variable de resistencia a la insulina, manteniendo la glucemia dentro de los límites normales (Abdul-Ghani and De- Fronzo 2009).
Esta observación es un argumento a favor de la hiperhormonemia pancreática como patognomónica de la diabetes. De ahí que la evaluación de los valores hormonales es un representante diagnóstico más específico y sensible en personas normoglucémicas, por lo demás sanas y no diabéticas según el enfoque diagnóstico glucocéntrico actual.
▶ De la hipótesis bihormonal a la disfunción multihormonal de los islotes
♦ Glucagón
El glucagón es una hormona segregada por las células alfa de los islotes pancreáticos con diversas funciones, entre ellas el mantenimiento de normoglucemia durante el ayuno. La disminución de la secreción de insulina y la secreción de glucagón debida a la estimulación con arginina se observa en personas con intolerancia a la glucosa años antes del diagnóstico (Larsson et al. 1995, Ahrén 2009). Estas características se observan en sujetos con glucosa y glucagón en sangre normales al inicio, pero que ya tenían mayor insulinemia en ayunas dentro del intervalo referencia normal.
♦ Polipéptido pancreático
El polipéptido pancreático (PP) es segregado por las células PP y está aumentado en las personas con DMT2. En ellas, la insulina, el PP y el glucagón aumentan durante la PTOG (Chia et al. 2014), lo que es una evidencia a favor de la hipótesis multihormonal.
♦ Amilina
La amilina es una hormona con 37 aminoácidos descubierta en1987, y es liberada junto con la insulina por los mismos gránulos secretorios de las células beta del páncreas (Guerreiro et al. 2013). La amilina es central para la regulación de la secreción del glucagón (Young, 2005).
En individuos normoglucémicos que progresan hacia la insuficiencia pancreática, se produce una hiperhormonemia paralela (hiperinsulinemia e hiperamilinemia), con aumento de la cantidad de amilina en relación con la de insulina en función del paso de tolerancia normal a la glucosa a intolerancia a la glucosa y por último DMT2 (Ludvik et al. 1991, Thomaseth et al. 1997, Kahn et al. 1998).
La insulina y la amilina son hormonas que pueden tener valores altos en ayunas, así como después de una carga de glucosa oral en algunas personas normoglucémicas no diabéticas, mientras que es más probable que el glucagón y el PP estén alterados en personas con diagnóstico de diabetes.
♦ Insulina
La insulinemia es muy variable entre los pacientes normoglucémicos sometidos a PTOG. En un estudio con personas sometidas a esta prueba se halló una dependencia bifásica de la respuesta insulinémica sobre la glucemia, primero con aumento brusco dentro de los límites normoglucémicos, que precedía a una fase de descenso en función del aumento de la glucemia dentro de los intervalos de intolerancia a la glucosa y diabéticos (Reaven et al.1967).
Kraft confirmó la respuesta hiperinsulinémica a una carga de glucosa oral durante una POTG prolongada – hasta 5 horas – en individuos normoglucémicos, lo que anticipa la disminución de la respuesta a la insulina observada en la diabetes manifiesta (Kraft 1975, 2008) (Fig. 1b). Los grados variables de respuesta insulinémica a la POTG – o incluso a las comidas – entre aquellos por lo demás considerados como personas sanas, brinda evidencia de los grados variables de resistencia a la insulina entre sujetos normoglucémicos.
Cuando se comparan grupos de adultos normoglucémicos no obesos y obesos, estos últimos muestran aumento de la insulinemia en ayunas y respuesta insulinémica de 24 hs, con escasa diferencia en el perfil glucémico de 24 hs (Polonsky et al. 1988). En un estudio con jóvenes normoglucémicos obesos (IMC ~ 35 kg/ cm2) el grupo era hiperinsulinémico (insulinemia en ayunas > 27 U/ml) y por lo tanto resistente a la insulina (HOMA-OR > 4,3) y también tuvo intolerancia a la glucosa con alto perfil insulinémico durante la POTG (Lustig et al. 2016).
Sin embargo, aún entre jóvenes delgados, normoglucémicos y normolipídicos (en ayunas) se puede hallar un subgrupo con aumento de la insulinemia en ayunas -todavía “normal” (< 13 mU/ml) – con perfil de 24 hs hiperinsulinémico y dislipidémico - en especial triglicéridos altos y nueva lipogénesis hepática (Petersen et al.2007) En el estudio San Antonio Metabolism (SAM), efectuado con 388 participantes obesos y no obesos (tolerancia a la glucosa normal, intolerancia a la glucosa y DMT2), la secreción de insulina durante la POTG mostró una forma típica en U invertida, mostrando la disminución de la secreción de insulina precedida por una fase hipersinsulinémica dentro de los intervalos normoglucémicos (Gastaldelli et al. 2004). También se halló que la disminución de la función de las células beta es un continuo en función de la glucemia en ayunas (o dela POTG de 2hs), que comienza con valores normoglucémicos.
Respuesta insulínica tras una carga oral de glucosa.
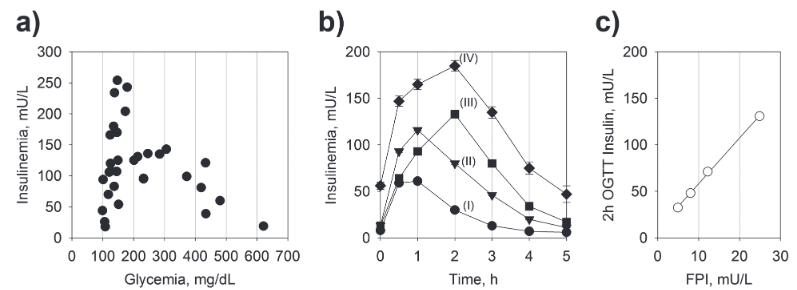
a) Respuesta insulinémica en individuos tras una carga oral de glucosa. Datos de (Reaven et al., 1967).
b) La respuesta insulínica en los individuos normoglucémicos después de una carga de glucosa oral. Cada curva (I, II, III y IV) representa un patrón (cuantil) con el promedio de las respuestas insulinémicas de un gran subconjunto de individuos que siguen una carga oral de glucosa. Datos de (Kraft 2008).
c) Correlación entre FPI y la insulinemia a las 2h después de una carga oral de glucosa. (Datos Johnson et al., 2010).
EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG
|
HOMA es una medición calculada a partir de los resultados de la glucemia y la insulinemia en ayunas durante los análisis habituales. HOMA tiene buen poder analítico, pero es una prueba compleja inadecuada para el diagnóstico clínico habitual. Aunque es adecuado para evaluar la función de las células beta y la resistencia a la insulina en personas con diagnóstico de prediabetes o diabetes, HOMA también puede ser evidencia directa de la función de las células beta en el intervalo de referencia normal. A fin de mantener un control estricto de la glucemia, las personas normoglucémicas tienen resistencia creciente a la insulina, que se manifiesta con insulinemia en aumento, en ayunas y en respuesta a una carga glucémica oral.
A partir de décadas de estudios independientes es evidente que se produce una disfunción multihormonal temprana como intento para normalizar la glucemia en el contexto de la degeneración metabólica. Más importante, este análisis muestra que la insulinemia en ayunas es un indicador directo y confiable de resistencia a la insulina en personas por lo demás no diabéticas y normoglucémicas.
La importancia del análisis de insulinemia en ayunas en la evaluación de la resistencia a la insulina antes del inicio de la diabetes clínica fue reconocida hace ya más de 20 años por la ADA.
El presente conjunto de datos indica que el enfoque glucocéntrico (glucemia en ayunas, HbA1c y POTG) no pronostica de manera terminante el riesgo a futuro de diabetes y sus complicaciones y comorbilidades. Dado que la resistencia a la insulina es la base subyacente de la intolerancia a la glucosa que genera el aumento de la glucemia y todos los demás marcadores para la diabetes y pre-diabetes y que la resistencia a la insulina se puede desarrollar aún en personas normoglucémicas, la evaluación directa de la resistencia a la insulina debe constituir el grupo de pruebas analíticas para la pesquisa de diabetes y se necesita únicamente la insulinemia en ayunas y la POTG confirmatorias con medición de la insulina.
Sensibilidad a la insulina en función de la insulinemia plasmática de acuerdo al modelo HOMA2

EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG
|
El espectro del hígado graso no alcohólico se consideró una manifestación hepática o un precursor del SMet (Smits et al. 2013, Lonardo et al. 2015. Además, se deben evaluar otros marcadores asociados al SMet ya que la acumulación de estos es indicativa de aumento de la resistencia a la insulina (Garg et al. 2011).
Estos marcadores del SMet son, entre otros, la grasa subcutánea, abdominal e intravisceral (IMC, índice cintura/cadera), parámetros cardiovasculares y ateroscleróticos (grosor de la íntima-media carotídea (Pais et al. 2016), calcio de la arteria coronaria - CAC, también llamado puntaje de calcio (Valenti et al. 2015)), presión arterial, inflamación y dislipidemia aterógena – aumento de los triglicéridoes, C-HDL bajo, apolipoproteína B aumentada o gran número de partículas de C-LDL (Leroux et al. 2000, Pourfarzib et al. 2014), numerosas partículas pequeñas de LDL y cLDL oxidado (Boizel et al. 2000).
Diagrama esquemático del continuo del riesgo de complicaciones comunes de la diabetes en función del metabolismo de la glucosa.

Marcadores: se muestra un gradiente de color continuo que va desde la parte inferior (verde, lado inferior izquierdo) hasta los riesgos más altos (rojo, superior derecho).
a) Representación esquemática de la progresión de riesgos (escala logarítmica) de futuras complicaciones y/o comorbilidades relacionadas a los marcadores clínicos oficiales de la diabetes adoptados actualmente (FPG y/o HbA1c y / o 2h-OGTT) y al rango normoglucémico (Diabetes subclínica). Diagrama inspirado en los estudios UKPDS (Stratton et al., 2000) y ARCS (Selvin et al., 2005a).
b) Progresión de complicaciones / comorbilidades de la diabetes en el rango subclínico (dentro de los rangos de referencia establecidos los síntomas de la diabetes) de los marcadores glucémicos y lipídicos. Estado del metabolismo de la glucosa para FPG, FPI / c-péptido / pro-insulina, HbA1c, glucosa 2h-OGTT. Estados de dislipidemia para TG, HDL, relación TG / HDL, oxLDL. Diagrama inspirado en el estudio de (Tirosh Et al. 2005).
c) Representación esquemática del comportamiento bifásico de la secreción de insulina en función de la progresión de la diabetes. A Bajos valores normoglicémicos de referencia de los indicadores de glucosa, la secreción de insulina se incrementa con el fin de compensar la construcción resistencia a la insulina. Diagrama inspirado en Reaven (Reaven et al., 1967), Kraft (Kraft 1975) y DeFronzo (Gastaldelli et al., 2004)
d) Representación esquemática de los marcadores clínicos asociados con hiperinsulinemia dentro de los rangos de referencia normales.
EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG
|
▶ Intervención en la prevención de la diabetes
◈ Dieta mediterránea
La dieta mediterránea fue eficaz para la prevención de la DMT2 en personas no diabéticas con alto riesgo de ECV, con un seguimiento medio de 5 años. Esta dieta es rica en verduras, aceite de oliva, pescado, frutas secas, fibra, polifenoles, ácido alfa-linoleico y productos naturales con procesamiento mínimo, baja en ácido linoleico y en alimentos ultraprocesados. (Salas-Salvadó et al. 2011, Estruch et al.2013, 2016, Babio et al. 2014).
El estudio Diabetes Prevention Program (DPP 2002) efectuado en grupos de alto riesgo mostró la reducción del 58 % del riesgo de diabetes mediante la intervención en los hábitos de vida. Incluyó la dieta del National Cholesterol Education Program Step 1 (NCEP Step 1), similar a la dieta mediterránea (KrisEtherton et al. 2001). Esta intervención demostró ser superior a la metformina (850 mg 2 veces al día.), ambas en relación con el placebo.
◈ Restricción calórica
Este enfoque se emplea desde hace más de 100 años, mucho antes de la insulina y otros fármacos para la diabetes (Mazur 2011). Estudios modernos confirmaron el protocolo de restricción calórica, la reversión de la disfunción orgánica y la normalización de los marcadores de diabetes (Steven et al. 2016). También la ADA lo reconoció como una intervención para mejorar la resistencia a la insulina (Association 1998).
Dado que el pico de la insulina postprandial puede tardar alrededor de 5 horas para llegar al valor basal (Gannon and Nuttall, 2004), se debe tener en cuenta la distribución de las comidas a lo largo del día, ya que es probable que impacte sobre el perfil insulinémico de 24 hs (Kahleova et al. 2014).
También se reconoce la gran variedad de la población en la repuesta glucémica a comidas similares (Zeevi et al. 2015) y la variabilidad de la respuesta glucémica individual a diferentes comidas con carga equivalente de macronutrientes.
◈ Restricción de hidratos de carbono/ carga glucémica
Si tomamos en cuenta la necesidad de disminuir el riesgo de diabetes y sus complicaciones a futuro, se debe considerar un enfoque de intervención en los hábitos de vida que disminuya las necesidades de insulina endógena, mejore la sensibilidad a la insulina, la función pancreática y los marcadores de riesgo más evidentes, como los triglicéridos, el C-HDL, la insulinemia y la glucemia.
Hay una relación directa entre la carga glucémica de los hidratos de carbono de la alimentación y las necesidades fisiológicas de insulina – el llamado índice insulínico de los alimentos (IIA). La carga de glucosa induce el aumento de glucemia e insulina, mientras la carga de otros macronutrientes no cambia significativamente ninguno de estos marcadores (Robertson et al. 2002, Bao et al. 2009).
Priorizar la preparación de comidas con bajo IIA, es decir con poca glucosa biodisponible del contenido total de hidratos de carbono como el almidón y los azúcares, junto con el empleo preferido de alimentos poco procesados (Canella et al. 2014) es una estrategia para disminuir la demanda excesiva sobre la función pancreática, superar el estado hiperinsulinémico prolongado, reducir la resistencia a la insulina y la aparición de parámetros relacionados con el SMet.
Hace tiempo que se conoce la importancia de la carga glucémica en el aumento a largo plazo de los triglicéridos (Reaven et al. 1967) y también está comprobado el efecto hipertrigliceridémico de la fructosa y su impacto sobre marcadores del SMet. Además, se planteó que la mayoría de las enfermedades metabólicas tienen más riesgo en condiciones de hiperinsulinemia (Cordain et al. 2003, Kopp 2003).
Se debatió mucho si los umbrales actuales para el diagnóstico de la diabetes deben disminuir y si la HbA1c y otras mediciones podrían formar un conjunto de análisis que evalúen mejor el riesgo de sufrir diabetes y sus complicaciones (Cefalu 2016, Yudkin 2016).
La extensa literatura científica citada en este trabajo expone evidencia sólida para la estricta dependencia de los marcadores de trastorno metabólico como un continuo de la insulinemia y como un indicador directo de la trayectoria de la resistencia a la insulina que tiene lugar en individuos normoglucémicos.
Es tiempo de reevaluar los criterios diagnósticos más allá del solo empleo de los marcadores glucocéntricos y de evaluar los niveles de resistencia a la insulina (a través de la insulinemia) y la manifestación hepática del síndrome metabólico (por medio del metabolismo de los lípidos, incluso dentro de la normolipemia), que comprende un conjunto de parámetros diagnósticos de marcadores directos de la base subyacente de la enfermedad.
El médico podría proponer intervenciones tempranas, no farmacológicas, sobre los hábitos de vida cuando se identifica un patrón de diabetes subclínica, a fin de mejorar la salud en general y disminuir los riesgos de diabetes a futuro y los riesgos evidentes de las complicaciones y comorbilidades relacionadas con la misma. Desde el punto de vista de la salud pública, adoptar políticas para promover alimentación y hábitos de vida saludables y la consiguiente reducción al mínimo de la carga de diabetes, complicaciones y comorbilidades, reduciría el impacto económico sobre el sistema de salud y la sociedad en general.
► Diabetes subclínica
- ABDUL-GHANI MA AND RA DEFRONZO. 2009. Plasma glucose concentration and prediction of future risk of type 2 diabetes. Diabetes Care 32(Suppl 2): S194-198.
- AHRÉN B. 2009. Beta- and alpha-cell dysfunction in subjects developing impaired glucose tolerance: outcome of a 12-year prospective study in postmenopausal Caucasian women. Diabetes 58: 726-731.
- ALBERTI KGMM ET AL. 2009. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 120:1640-1645.
- AMES BN. 2006. Low micronutrient intake may accelerate the degenerative diseases of aging through allocation of scarce micronutrients by triage. Proc Natl Acad Sci U S A 103: 17589-17594.
- ASSOCIATION AD. 1998. Consensus Development Conference on Insulin Resistance: 5-6 November 1997. Diabetes Care 21: 310-314.
- ASSOCIATION AD. 2014. Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 37: S81-S90.
- ASSOCIATION AD. 2016. 2. Classification and Diagnosis of Diabetes. Diabetes Care 39: S13-S22.
- ATKINSON FS, FOSTER-POWELL K AND BRANDMILLER JC. 2008. International Tables of Glycemic Index and Glycemic Load Values: 2008. Diabetes Care 31: 2281-2283.
- BABIO N ET AL. 2014. Mediterranean diets and metabolic syndrome status in the PREDIMED randomized trial. CMAJ 186: E649-657.
- BAO J, ATKINSON F, PETOCZ P, WILLETT WC AND BRAND-MILLER JC. 2011. Prediction of postprandial glycemia and insulinemia in lean, young, healthy adults: glycemic load compared with carbohydrate content alone. Am J Clin Nutr 93: 984-996.
- BAO J, DE JONG V, ATKINSON F, PETOCZ P AND BRANDMILLER JC. 2009. Food insulin index: physiologic basis for predicting insulin demand evoked by composite meals. Am J Clin Nutr 90: 986-992.
- BARR ELM, BOYKO EJ, ZIMMET PZ, WOLFE R, TONKIN AM AND SHAW JE. 2009. Continuous relationships between non-diabetic hyperglycaemia and both cardiovascular disease and all-cause mortality: the Australian Diabetes, Obesity, and Lifestyle (AusDiab) study. Diabetologia 52: 415-424.
- BARR ELM ET Al. 2010. HOMA insulin sensitivity index and the risk of all-cause mortality and cardiovascular disease events in the general population: the Australian Diabetes, Obesity and Lifestyle Study (AusDiab) study. Diabetologia 53: 79-88.
- BASH LD, SELVIN E, STEFFES M, CORESH J AND ASTOR BC. 2008. Poor glycemic control in diabetes and the risk of incident chronic kidney disease even in the absence of albuminuria and retinopathy: Atherosclerosis risk in communities (aric) study. Arch Intern Med 168:2440-2447.
- BOIZEL R, BENHAMOU PY, LARDY B, LAPORTE F, FOULON T AND HALIMI S. 2000. Ratio of triglycerides to HDL cholesterol is an indicator of LDL particle size in patients with type 2 diabetes and normal HDL cholesterol levels. Diabetes Care 23: 1679-1685.
- BONORA E ET AL. 2007. Insulin resistance as estimated by homeostasis model assessment predicts incident symptomatic cardiovascular disease in caucasian subjects from the general population: the Bruneck study. Diabetes Care 30:318-324.
- BOWER RL AND DL HAY. 2016. Amylin structure-function relationships and receptor pharmacology: implications for amylin mimetic drug development. Br J Pharmacol 173:1883-1898.
- BOZZETTO L ET AL. 2016. Extra-Virgin Olive Oil Reduces Glycemic Response to a High-Glycemic Index Meal in Patients With Type 1 Diabetes: A Randomized Controlled Trial. Diabetes Care 39: 518-524.
- BRUDEVOLD R, HOLE T AND HAMMERSTRØM J. 2008. Hyperferritinemia Is Associated with Insulin Resistance and Fatty Liver in Patients without Iron Overload. PLOS ONE 3: e3547.
- BUDOFF MJ ET AL. 2013. Progression of Coronary Calcium and Incident Coronary Heart Disease Events: MESA (Multi-Ethnic Study of Atherosclerosis). J Am Coll Cardiol 61: 1231-1239.
- BUTLER AE, JANSON J, BONNER-WEIR S, RITZEL R, RIZZA RA AND BUTLER PC. 2003. beta-Cell Deficit and Increased beta-Cell Apoptosis in Humans With Type 2
- Diabetes. Diabetes 52: 102-110.
- CABRERA DE LEÓN A ET AL. 2015. C-peptide as a risk factor of coronary artery disease in the general population. Diab Vasc Dis Res 12: 199-207.
- CAHILL GF. 2006. Fuel metabolism in starvation. Annu Rev Nutr 26: 1-22.
- CANELLA DS ET AL. 2014. Ultra-Processed Food Products and Obesity in Brazilian Households (2008-2009). PLOS ONE 9: e92752.
- CEFALU WT. 2016. “Prediabetes”: Are There Problems With This Label? No, We Need Heightened Awareness of This Condition! Diabetes Care 39: 1472-1477.
- CHIA CW, ODETUNDE JO, KIM W, CARLSONOD, FERRUCCI L AND EGAN JM. 2014. GIP Contributes to Islet Trihormonal Abnormalities in Type 2 Diabetes. J Clin
- Endocrinol Metab 99: 2477-2485.
- COLAGIURI S ET AL. 2011. Glycemic Thresholds for Diabetes-Specific Retinopathy Implications for diagnostic criteria for diabetes. Diabetes Care 34: 145-150.
- COOPER GJ, WILLIS AC, CLARK A, TURNER RC, SIM RB AND REID KB. 1987. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc Natl Acad Sci U S A 84: 8628-8632.
- CORDAIN L, EADES MR AND EADES MD. 2003. Hyperinsulinemic diseases of civilization: more than just Syndrome X. Comp Biochem Physiol A Mol Integr Physiol 136: 95-112.
- CRANE PK ET AL. 2013. Glucose Levels and Risk of Dementia. N Engl J Med 369: 540-548.
- CUNNANE SC AND CRAWFORD MA. 2003. Survival of the fattest: fat babies were the key to evolution of the large human brain. Comp Biochem Physiol A Mol Integr Physiol 136: 17-26.
- DABELEA D ET AL. 2014. PRevalence of type 1 and type 2 diabetes among children and adolescents from 2001 to 2009. JAMA 311: 1778-1786.
- DCCT. 1993. The Effect of Intensive Treatment of Diabetes on the Development and Progression of Long-Term Complications in Insulin-Dependent Diabetes Mellitus. N Engl J Med 329: 977-986.
- DCCT-EDIC. 2000. Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. N Engl J Med 342: 381-389.
- DECODE STUDY GROUP. 1999. Glucose tolerance and mortality: comparison of WHO and American Diabetic Association diagnostic criteria. The DECODE study group. European Diabetes Epidemiology Group. Diabetes Epidemiology: Collaborative analysis Of Diagnostic criteria in Europe. The Lancet 354: 617-621.
- DECODE STUDY GROUP, EUROPEAN DIABETES EPIDEMIOLOGY GROUP. 2003. Is the current definition for diabetes relevant to mortality risk from all causes and cardiovascular and noncardiovascular diseases? Diabetes Care 26: 688-696.
- DEFRONZO RA. 1988. Lilly lecture 1987. The triumvirate: beta-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes 37: 667-687.
- DEFRONZO RA, TOBIN JD AND ANDRES R. 1979. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 237: E214-223.
- DESPRÉS JP ET AL. 1996. Hyperinsulinemia as an independent risk factor for ischemic heart disease. N Engl J Med 334 :952-957.
- DEVECI E ET AL. 2009. Evaluation of insulin resistance in normoglycemic patients with coronary artery disease. Clin Cardiol 32: 32-36.
- DEWITT DE AND IB HIRSCH. 2003. Outpatient insulin therapy in type 1 and type 2 diabetes mellitus: scientific review. JAMA 289:2254-2264.
- DOBBS R ET AL. 1975. Glucagon: role in the hyperglycemia of diabetes mellitus. Science 187: 544-547.
- DPP - DIABETES PREVENTION PROGRAM. 2002. Reduction in the Incidence of Type 2 Diabetes with Lifestyle Intervention or Metformin. Diabetes Prevention Program Research Group. N Engl J Med 346: 393-403.
- EBBELING CB, LEIDIG MM, FELDMAN HA, LOVESKY MM AND LUDWIG DS. 2007. Effects of a low-glycemic load vs low-fat diet in obese young adults: a randomized trial. JAMA 297: 2092-2102.
- ESTRUCH R ET AL. 2016. Effect of a high-fat Mediterranean diet on bodyweight and waist circumference: a prespecified secondary outcomes analysis of the PREDIMED randomised controlled trial. Lancet Diabetes Endocrinol 4: 666-676.
- ESTRUCH R ET AL. 2013. Primary Prevention of Cardiovascular Disease with a Mediterranean Diet. research-article.
- FEDERATION. 2015. IDF Diabetes Atlas. P. In: International Diabetes Federation. FEINMAN RD ET AL. 2015. Dietary carbohydrate restriction as the first approach in diabetes management: critical review and evidence base. Nutr Burbank Los Angel Cty Calif 31: 1-13.
- FLOYD JC, FAJANS SS, PEK S AND CHANCE RE. 1976. A newly recognized pancreatic polypeptide; plasma levels in health and disease. Recent Prog Horm Res 33: 519-570.
- FORGA LLENAS L, GOÑI IRIARTE MJ, CAMBRA CONTIN K, IBÁÑEZ BEROIZ B, CHUECA GUENDULAIN M AND BERRADE ZUBIRI S. 2015. Incidence and temporal trends of childhood type 1 diabetes between 1975 and 2012 in Navarre (Spain). Gac Sanit SESPAS 29: 51-54.
- FOROUHI NG AND WAREHAM NJ. 2014. Epidemiology of diabetes. Medicine (Baltimore) 42: 698-702.
- GANNON MC AND NUTTALL FQ. 2004. Effect of a highprotein, low-carbohydrate diet on blood glucose control in people with type 2 diabetes. Diabetes 53: 2375-2382.
- GARBER AJ ET AL. 2016. Consensus statement by the american association of clinical endocrinologists and american college of endocrinology on the comprehensive type 2 diabetes management algorithm - 2016 executive summary. Endocr Pract 22: 84-113.
- GARDNER CD ET AL. 2007. Comparison of the Atkins, Zone, Ornish, and LEARN diets for change in weight and related risk factors among overweight premenopausal women: the A TO Z Weight Loss Study: a randomized trial. JAMA 297: 969-977.
- GARG MK, DUTTA MK and MAHALLE N. 2011. Study of beta-cell function (by HOMA model) in metabolic syndrome. Indian J Endocrinol Metab 15: S44-49.
- GASTALDELLI A, FERRANNINI E, MIYAZAKI Y, MATSUDA M, DEFRONZO RA and SAN ANTONIO METABOLISM STUDY. 2004. Beta-cell dysfunction and glucose intolerance: results from the San Antonio metabolism (SAM) study. Diabetologia 47: 31-39.
- GODOY-MATOS AF. 2014. The role of glucagon on type 2 diabetes at a glance. Diabetol Metab Syndr 6: 91.
- GOTO A ET AL. 2016. High hemoglobin A1c levels within the non-diabetic range are associated with the risk of all cancers. Int J Cancer 138: 1741-1753.
- GRUNDY SM ET AL. 2005. Diagnosis and Management of the Metabolic Syndrome. Circulation 112: 2735-2752.
- GUERREIRO LH, DA SILVA D, SOLA-PENNA M, MIZURINI DM and LIMA LMTR. 2013. Amylin induces hypoglycemia in mice. An Acad Bras Cienc 85: 349-354.
- HALL KD ET AL. 2015. Calorie for Calorie, Dietary Fat Restriction Results in More Body Fat Loss than Carbohydrate Restriction in People with Obesity. Cell Metab 22: 427-436.
- HANLEY AJG, WILLIAMS K, STERN MP AND HAFFNER SM. 2002. Homeostasis model assessment of insulin resistance in relation to the incidence of cardiovascular disease: the San Antonio Heart Study. Diabetes Care 25:1177-1184.
- HARJUTSALO V, SJÖBERG L AND TUOMILEHTO J. 2008. Time trends in the incidence of type 1 diabetes in Finnish children: a cohort study. The Lancet 371: 1777-1782.
- HÄTÖNEN KA ET AL. 2006. Methodologic considerations in the measurement of glycemic index: glycemic response to rye bread, oatmeal porridge, and mashed potato. Am J Clin Nutr 84: 1055-1061.
- HAY DL, CHEN S, LUTZ TA, PARKES DG AND ROTH JD. 2015. Amylin: Pharmacology, Physiology, and Clinical Potential. Pharmacol Rev 67: 564-600. HECHT HS. 2015. Coronary artery calcium scanning: past, present, and future. JACC Cardiovasc. Imaging 8: 579-596.
- HOLDEN SE ET AL. 2013. The incidence of type 2 diabetes in the United Kingdom from 1991 to 2010. Diabetes Obes Metab 15: 844-852.
- HUO X ET AL. 2016. Risk of non-fatal cardiovascular diseases in early-onset versus late-onset type 2 diabetes in China: a cross-sectional study. Lancet Diabetes Endocrinol 4: 115-124.
- IKEDA F ET AL. 2013. Haemoglobin A1c even within nondiabetic level is a predictor of cardiovascular disease in a general Japanese population: the Hisayama Study. Cardiovasc Diabetol 12: 164.
- INSTITUTE OF MEDICINE. 2005. Dietary Reference Intakes for Energy, Carbohydrate, Fiber, Fat, Fatty Acids, Cholesterol, Protein, and Amino Acids (Macronutrients). P. in.: National Academies Press, Washington, D.C.
- INSTITUTE OF MEDICINE. 2006. Dietary Reference Intakes Research Synthesis: Workshop Summary. P. In: National Academies Press, Washington, D.C.
- JENKINS DJ ET AL. 1981. Glycemic index of foods: a physiological basis for carbohydrate exchange. Am J Clin Nutr 34: 362-366.
- JOHNSON JL, DUICK DS, CHUI MA and ALDASOUQI SA. 2010. Identifying prediabetes using fasting insulin levels. Endocr Pract 16: 47-52.
- JONES AG AND HATTERSLEY AT. 2013. The clinical utility of C-peptide measurement in the care of patients with diabetes. Diabet Med 30: 803-817.
- KAHLEOVA H ET AL. 2014. Eating two larger meals a day (breakfast and lunch) is more effective than six smaller meals in a reduced-energy regimen for patients with type 2 diabetes: a randomised crossover study. Diabetologia 57:1552-1560.
- KAHN SE ET AL. 1998. Reduced amylin release is a characteristic of impaired glucose tolerance and type 2 diabetes in Japanese Americans. Diabetes 47: 640-645.
- KARROWNI W ET AL. 2013. Insulin resistance is associated with significant clinical atherosclerosis in nondiabetic patients with acute myocardial infarction. Arterioscler Thromb Vasc Biol 33: 2245-2251.
- KATO M, NODA M, SUGA H, MATSUMOTO M AND KANAZAWA Y. 2009. Fasting plasma glucose and incidence of diabetes --- implication for the threshold for impaired fasting glucose: results from the populationbased Omiya MA cohort study. J Atheroscler Thromb 16:857-861.
- KAUR B AND HENRY J. 2014. Micronutrient status in type 2 diabetes: a review. Adv Food Nutr Res 71: 55-100.
- KHAW KT, WAREHAM N, BINGHAM S, LUBEN R, WELCH A AND DAY N. 2004. Association of hemoglobin A1c with cardiovascular disease and mortality in adults:the European prospective investigation into cancer in Norfolk. Ann Intern Med 141: 413-420.
- KOHNER EM ET AL. 1998. United Kingdom Prospective Diabetes Study, 30: diabetic retinopathy at diagnosis of non-insulin-dependent diabetes mellitus and associated risk factors. Arch Ophthalmol 116(3): 297-303.
- KOPP W. 2003. High-insulinogenic nutrition--an etiologic factor for obesity and the metabolic syndrome? Metabolism 52: 840-844.
- KRAFT JR. 1975. Detection of Diabetes Mellitus In Situ (Occult Diabetes). Lab Med 6: 10-22.
- KRAFT JR. 2008. Diabetes Epidemic & You. P. In.: Trafford Publishing.
- KRIS-ETHERTON P, ECKEL RH, HOWARD BV, JEOR SS AND BAZZARRE TL. 2001. Lyon Diet Heart Study. Circulation 103: 1823-1825.
- LAAKSO M. 1993. How good a marker is insulin level for insulin resistance? Am J Epidemiol 137: 959-965.
- LAMB MM, FREDERIKSEN B, SEIFERT JA, KROEHL M, REWERS M AND NORRIS JM. 2015. Sugar intake is associated with progression from islet autoimmunity to type 1 diabetes: the Diabetes Autoimmunity Study in the Young. Diabetologia 58: 2027-2034.
- LARSSON H, BERGLUND G AND AHRÉN B. 1995. Glucose modulation of insulin and glucagon secretion is altered in impaired glucose tolerance. J Clin Endocrinol Metab 80: 1778-1782.
- LEEMAN M, OSTMAN E AND BJÖRCK I. 2005. Vinegar dressing and cold storage of potatoes lowers postprandial glycaemic and insulinaemic responses in healthy subjects. Eur J Clin Nutr 59: 1266-1271.
- LEROUX G ET AL. 2000. Influence of triglyceride concentration on the relationship between lipoprotein cholesterol and apolipoprotein B and A-I levels. Metabolism 49: 53-61.
- LI Y, LI Y, MENG L AND ZHENG L. 2015. Association between Serum C-Peptide as a Risk Factor for Cardiovascular Disease and High-Density Lipoprotein Cholesterol Levels in Nondiabetic Individuals. PLoS ONE 10.
- LIM JS, MIETUS-SNYDER M, VALENTE A, SCHWARZ JM AND LUSTIG RH. 2010. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat Rev Gastroenterol Hepatol 7: 251-264.
- LIMA LMTR. 2017. Prediabetes definitions and clinical outcomes. Lancet Diabetes Endocrinol 5: 92-93.
- LINDAHL B, DINESEN B, ELIASSON M, RØDER M, HALLMANS G AND STEGMAYR B. 2000. High Proinsulin Levels Precede First-Ever Stroke in a Nondiabetic Population. Stroke 31: 2936-2941.
- LONARDO A, BALLESTRI S, MARCHESINI G, ANGULO P AND LORIA P. 2015. Nonalcoholic fatty liver disease: A precursor of the metabolic syndrome. Dig Liver Dis 47:181-190.
- LOOKER HC ET AL. 2012. Diabetic retinopathy at diagnosis of type 2 diabetes in Scotland. Diabetologia 55: 2335-2342.
- LUCHSINGER JA TANG MX, SHEA S AND MAYEUX R. 2004. Hyperinsulinemia and risk of Alzheimer disease. Neurology 63: 1187-1192.
- LUDVIK B, LELL B, HARTTER E, SCHNACK C and PRAGER R. 1991. Decrease of stimulated amylin release precedes impairment of insulin secretion in type II diabetes. Diabetes 40: 1615-1619.
- LUSTIG RH ET AL. 2016. Isocaloric fructose restriction and metabolic improvement in children with obesity and metabolic syndrome. Obes Silver Spring Md 24: 453-460.
- MARLISS EB, AOKI TT, UNGER RH, SOELDNER JS AND CAHILL GF. 1970. Glucagon levels and metabolic effects in fasting man. J Clin Invest 49: 2256-2270.
- MATTHEWS DR, JP HOSKER, AS RUDENSKI, BA NAYLOR, DF TREACHER AND RC TURNER. 1985. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28: 412-419.
- MAZUR A. 2011. Why were “starvation diets” promoted for diabetes in the pre-insulin period? Nutr J 10: 23.
- MCCLAIN AD, OTTEN JJ, HEKLER EB AND GARDNER CD. 2013. Adherence to a low-fat vs. low-carbohydrate diet differs by insulin resistance status. Diabetes Obes Metab 15: 87-90.
- MECHANICK JI. 2015. Global Dimensions of Diabetes: Information and Synthesis. Ann Glob Health 81: 733-734.
- MENKE A, CASAGRANDE S, GEISS L AND COWIE CC. 2015. Prevalence of and trends in diabetes among adults in the United States, 1988-2012. JAMA 314: 1021-1029.
- MIAO X, SUN W, FU Y, MIAO L AND CAI L. 2013. Zinc homeostasis in the metabolic syndrome and diabetes. Front Med 7: 31-52.
- MOZAFFARIAN D AND JHY WU. 2012. (n-3) fatty acids and cardiovascular health: are effects of EPA and DHA shared or complementary? J Nutr 142: 614S-625S.
- MUTI P ET AL. 2002. Fasting glucose is a risk factor for breast cancer: a prospective study. Cancer Epidemiol Biomarkers Prev 11: 1361-1368.
- NAGI DK, PETTITT DJ, BENNETT PH, KLEIN R AND KNOWLER WC. 1997. Diabetic retinopathy assessed by fundus photography in Pima Indians with impaired glucose tolerance and NIDDM. Diabet Med J Br Diabet Assoc 14: 449-456.
- NATHAN DM ET AL. 2005. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 353:2643-2653.
- NCD-RISC. 2016. Worldwide trends in diabetes since 1980:a pooled analysis of 751 population-based studies with 4·4 million participants. NCD Risk Factor Collaboration (NCD-RisC). The Lancet 387: 1513-1530.
- NICHOLS GA, HILLIER TA and BROWN JB. 2007. Progression from newly acquired impaired fasting glucose to type 2 diabetes. Diabetes Care 30: 228-233.
- NICHOLS GA, HILLIER TA and BROWN JB. 2008. Normal fasting plasma glucose and risk of type 2 diabetes diagnosis. Am J Med 121: 519-524.
- OHN JH ET AL. 2016. 10-year trajectory of beta-cell function and insulin sensitivity in the development of type 2 diabetes: a community-based prospective cohort study. Lancet Diabetes Endocrinol 4: 27-34.
- ONIKI K ET AL. 2016. The longitudinal effect of the aldehyde dehydrogenase 2*2 allele on the risk for nonalcoholic fatty liver disease. Nutr Diabetes 6: e210.
- OSTMAN E, GRANFELDT Y, PERSSON L AND BJÖRCK I. 2005. Vinegar supplementation lowers glucose and insulin responses and increases satiety after a bread meal in healthy subjects. Eur J Clin Nutr 59: 983-988.
- PAIS R ET AL. 2016. Fatty liver is an independent predictor of early carotid atherosclerosis. J Hepatol 65(1): 95-102.
- PATTERSON CC, DAHLQUIST GG, GYÜRÜS E, GREEN A AND SOLTÉSZ G. 2009. Incidence trends for childhood type 1 diabetes in Europe during 1989-2003 and predicted new cases 2005-20: a multicentre prospective registration study. The Lancet 373: 2027-2033.
- PETERSEN KF ET AL. 2007. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci U S A 104: 12587-12594.
- POLONSKY KS, GIVEN BD AND VAN CAUTER E. 1988. Twenty-four-hour profiles and pulsatile patterns of insulin secretion in normal and obese subjects. J Clin Invest 81:442-448.
- POURFARZIB R ET AL. 2014. Relationship between plasma apolipoprotein B concentrations and LDL particle number. Research Reports in Clinical Cardiology.
- REAVEN GM, FARQUHAR JW AND NAKANISHI RH. 1969. Steady State Plasma Insulin Response to Continuous Glucose Infusion in Normal and Diabetic Subjects. Diabetes 18: 273-279.
- REAVEN GM, LERNER RL, STERN MP AND FARQUHAR JW. 1967. Role of insulin in endogenous hypertriglyceridemia. J Clin Invest 46: 1756-1767.
- ROBERTSON MD, HENDERSON RA, VIST GE and RUMSEY RDE. 2002. Extended effects of evening meal carbohydrate-to-fat ratio on fasting and postprandial substrate metabolism. Am J Clin Nutr 75: 505-510.
- RYDÉN L ET AL. 2007. Guidelines on diabetes, pre-diabetes, and cardiovascular diseases: executive summary. Eur Heart J 28: 88-136.
- RYDÉN L, VIVECA G, SCHNELL O and JAAKKO T. 2016. Oral glucose tolerance testing and cardiovascular disease. Lancet Diabetes Endocrinol 4: 732-733.
- SALAS-SALVADÓ J ET AL. 2011. Reduction in the Incidence of Type 2 Diabetes With the Mediterranean Diet. Diabetes Care 34: 14-19.
- SANYAL D, MUKHERJEE P, RAYCHAUDHURI M, GHOSH S, MUKHERJEE S and CHOWDHURY S. 2015. Profile of liver enzymes in non-alcoholic fatty liver disease in patients with impaired glucose tolerance and newly detected untreated type 2 diabetes. Indian J Endocrinol Metab 19: 597-601.
- SCHMIDT MI ET AL. 2011. Chronic non-communicable diseases in Brazil: burden and current challenges. The Lancet 377: 1949-1961.
- SCHWARTZ SS, EPSTEIN S, CORKEY BE, GRANT SFA, GAVIN JR AND AGUILAR RB. 2016. The Time Is Right for a New Classification System for Diabetes: Rationale and Implications of the beta-Cell-Centric Classification Schema. Diabetes Care 39: 179-186.
- SCHWARZ JM ET AL. 2015. Effect of a High-Fructose Weight-Maintaining Diet on Lipogenesis and Liver Fat. J Clin Endocrinol Metab 100: 2434-2442.
- SELVIN E, CORESH J, GOLDEN SH, BRANCATI FL, FOLSOM AR AND STEFFES MW. 2005a. Glycemic Control and Coronary Heart Disease Risk in Persons With and Without Diabetes: The Atherosclerosis Risk in Communities Study. Arch Intern Med 165: 1910.
- SELVIN E, CORESH J, SHAHAR E, ZHANG L, STEFFES M AND SHARRETT AR. 2005b. Glycaemia (haemoglobin A1c) and incident ischaemic stroke: the Atherosclerosis Risk in Communities (ARIC) Study. Lancet Neurol 4: 821-826.
- SELVIN E ET AL. 2010. Glycated hemoglobin, diabetes, and cardiovascular risk in nondiabetic adults. N Engl J Med 362: 800-811.
- SHALAUROVA I, CONNELLY MA, GARVEY WT AND OTVOS JD. 2014. Lipoprotein insulin resistance index: a lipoprotein particle-derived measure of insulin resistance. Metab Syndr Relat Disord 12: 422-429.
- SHAW JE ET AL. 2000. Impaired fasting glucose: how low should it go? Diabetes Care 23: 34-39.
- SIERI S ET AL. 2012. Prospective study on the role of glucose metabolism in breast cancer occurrence. Int J Cancer 130:921-929.
- SIMMONS D AND HLAING T. 2014. Interpretation of HbA1c: association with mean cell volume and haemoglobin concentration. Diabet Med 31: 1387-1392.
- SISNANDE T ET AL. 2015. Monoconjugation of Human Amylin with Methylpolyethyleneglycol. PloS One 10:e0138803.
- SMITS MM, IOANNOU GN, BOYKO EJ and UTZSCHNEIDER KM. 2013. Non-alcoholic fatty liver disease as an independent manifestation of the metabolic syndrome: results of a US national survey in three ethnic groups. J Gastroenterol Hepatol 28: 664-670.
- SONG SH. 2016. Early-onset type 2 diabetes: high lifetime risk for cardiovascular disease. Lancet Diabetes Endocrinol 4:87-88.
- STEVEN S ET AL. 2016. Very-Low-Calorie Diet and 6 Months of Weight Stability in Type 2 Diabetes: Pathophysiologic Changes in Responders and Nonresponders. Diabetes Care 39(5): 808-815.
- STRATTON IM ET AL. 2000. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ 321: 405-412.
- THOMASETH K ET AL. 1997. Amylin release during oral glucose tolerance test. Diabet Med J Br Diabet Assoc 14(Suppl 2): S29-34.
- TIROSH A ET AL. 2005. Normal fasting plasma glucose levels and type 2 diabetes in young men. N Engl J Med 353: 1454-1462.
- TOSCANO CM ET AL. 2015. Cost-effectiveness of a national population-based screening program for type 2 diabetes: the Brazil experience. Diabetol Metab Syndr 7: 95.
- TÓTH C AND CLEMENS Z. 2015. A child with type 1 diabetes mellitus (T1DM) successfully treated with the Paleolithic ketogenic diet: A 19-month insulin-freedom. Int J Case Rep Images 6: 752.
- UKPDS. 1998a. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet Lond Engl 352: 854-865.
- UKPDS. 1998b. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet Lond Engl 352: 837-853.
- UNGER RH AND ORCI L. 1975. The essential role of glucagon in the pathogenesis of diabetes mellitus. Lancet Lond Engl 1:14-16.
- UNWIN DJ, CUTHBERTSON DJ, FEINMAN RD and SPRUNG VS. 2015. A pilot study to explore the role of a low-carbohydrate intervention to improve GGT levels and HbA1c. Diabesity in Practice 4: 102-108. USDA. 2014. Nutrient Data : SR27. USDA - United States Department of Agriculture, ARS - Agricultural Research Service.
- VALENTI V ET AL. 2015. A 15-Year Warranty Period for Asymptomatic Individuals Without Coronary Artery Calcium: A Prospective Follow-Up of 9,715 Individuals. JACC Cardiovasc Imaging 8: 900-909.
- VALENTI V ET AL. 2016. Absence of Coronary Artery Calcium Identifies Asymptomatic Diabetic Individuals at Low Near-Term But Not Long-Term Risk of Mortality A 15-Year Follow-Up Study of 9715 Patients. Circ Cardiovasc Imaging 9: e003528.
- VAN’T RIET E ET AL. 2012. HbA1c is an independent predictor of non-fatal cardiovascular disease in a Caucasian population without diabetes: a 10-year follow-up of the Hoorn Study. Eur J Prev Cardiol 19: 23-31.
- WALLACE TM, LEVY JC AND MATTHEWS DR. 2004. Use and Abuse of HOMA Modeling. Diabetes Care 27:1487-1495.
- WEISS R, BREMER AA AND LUSTIG RH. 2013. What is metabolic syndrome, and why are children getting it? Ann N Y Acad Sci 1281: 123-140.
- WESTERMARK P, WERNSTEDT C, WILANDER E, HAYDEN DW, O’BRIEN TD and JOHNSON KH. 1987. Amyloid fibrils in human insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc Natl Acad Sci U S A 84: 3881-3885.
- WHO. 2015. Health topics - Diabetes. World Health Organizaiton (WHO). WHO.
- WHO. 2016. Use of glycated haemoglobin (HbA1c) in the diagnosis of diabetes mellitus. World Health Organization (WHO). WHO.
- WIENER K. 1995. Fasting plasma glucose as a diagnostic indicator of diabetes mellitus. Clin Chim Acta Int J Clin Chem 238: 199-208.
- WONG ND ET AL. 2003. The metabolic syndrome, diabetes, and subclinical atherosclerosis assessed by coronary calcium. J Am Coll Cardiol 41: 1547-1553.
- WONG ND ET AL. 2012. Metabolic Syndrome, Diabetes, and Incidence and Progression of Coronary Calcium: The Multiethnic Study of Atherosclerosis Study. JACC Cardiovasc Imaging 5: 358-366.
- WU A. 2006. Tietz Clinical Guide to Laboratory Tests Fourth Edition. P. In.: Saunders, St. Louis, Mo.
- YALOW RS AND SA BERSON. 1960. Immunoassay of endogenous plasma insulin in man. J. Clin. Invest. 39: 1157-1175.
- YALOW RS, GLICK SM, ROTH J AND BERSON SA. 1965. Plasma insulin and growth hormone levels in obesity and diabetes. Ann N Y Acad Sci 131: 357-373.
- YOUNG A. 2005. Inhibition of glucagon secretion. Adv Pharmacol San Diego Calif 52: 151-171.
- YUDKIN JS. 2016. “Prediabetes”: Are There Problems With This Label? Yes, the Label Creates Further Problems! Diabetes Care 39: 1468-1471.
- YUDKIN JS AND MONTORI VM. 2014. The epidemic of pre-diabetes: the medicine and the politics. BMJ 349:g4485.
- ZEEVI D ET AL. 2015. Personalized Nutrition by Prediction of Glycemic Responses. Cell 163: 1079-1094.
- ZHANG P, ENGELGAU MM, VALDEZ R, CADWELL B, BENJAMIN SM and NARAYAN KMV. 2005. Efficient cutoff points for three screening tests for detecting undiagnosed diabetes and pre-diabetes: an economic analysis. Diabetes Care 28: 1321-1325.
- ZILKER TR ET AL. 1988. Pharmacokinetics of biosynthetic human proinsulin following intravenous and subcutaneous administration in metabolically healthy volunteers. Horm Metab Res Suppl Ser 18: 37-43.

No hay comentarios:
Publicar un comentario