
Fibrosis pulmonar idiopática y determinantes genéticos
Actualización y revisión en profundidad: Enfermedad pulmonar crónica, progresiva, irreversible y letal, de causa desconocida. Estudios genéticos ofrecen la esperanza de identificar con más precisión a individuos en riesgo de padecerla; de definir su patogenia y de crear futuros tratamientos dirigidos
Actualización y revisión en profundidad: Enfermedad pulmonar crónica, progresiva, irreversible y letal, de causa desconocida. Estudios genéticos ofrecen la esperanza de identificar con más precisión a individuos en riesgo de padecerla; de definir su patogenia y de crear futuros tratamientos dirigidos
Autor(es): Dres. Spagnolo P, Grunewald J, du Bois RM y Dres. Talmadage E King Jr, Annie Pardo, Moisés Selman

|
La fibrosis pulmonar idiopática (FPI), la forma más común de las neumonías intersticiales idiopáticas, es una enfermedad pulmonar crónica, progresiva, irreversible y letal, de causa desconocida. Se produce en personas de mediana edad y adultos de edad avanzada (edad media de diagnóstico 66 años, se limita a los pulmones y se asocia con un patrón histopatológico o radiológico típico de neumonía intersticial usual (NIU).
La característica histopatológica principal de la NIU, mejor vista con poco aumento, es el aspecto heterogéneo. Se observan áreas de fibrosis subpleural y paraseptal y panal de abejas (espacios aéreos de fibrosis quística revestidos por epitelio bronquiolar a menudo llenos de mucina y, un número variable de células inflamatorias) que alternan con zonas de parénquima menos afectado o normal (heterogeneidad espacial). En el fondo de los depósitos de colágeno se hallan pequeñas áreas de fibrosis activa (focos de fibroblastos) que reflejan la heterogeneidad temporal del proceso patológico e indican las actividades actuales.
En general, la inflamación es leve y consiste en un infiltrado intersticial linfoplasmocitario desigual. Los signos de la tomografía computarizada (TC) de alta resolución representan el patrón de la NIU, caracterizado por opacidades reticulares a menudo asociadas con bronquiectasias de tracción, con poca o ninguna opacificación en vidrio esmerilado. Es común la presencia de lesiones subpleurales en panal de abejas, con espacios aéreos quísticos agrupados con paredes bien definidas (por lo general de 3-10 mm de diámetro). La presencia de estas lesiones es fundamental para hacer un diagnóstico definitivo.
Los pacientes con FPI generalmente solicitan atención médica porque sufren disnea de esfuerzo y tos crónicas y progresivas. La auscultación pulmonar halla con frecuencia estertores crepitantes inspiratorios bibasales; se constatan dedos en palillo de tambor. La historia natural de la FPI se caracteriza por ser un trastorno pulmonar estable o lentamente progresivo, patrón que siguen la mayoría de los pacientes. Sin embargo, los últimos hallazgos indican que la FPI es un grupo heterogéneo enfermedades y se están describiendo nuevos fenotipos clínicos con distintos patrones de supervivencia.
Los mecanismos patogénicos no están claros, pero cada vez hay más evidencia que indica que la enfermedad es el resultado de un comportamiento anormal de las células del epitelio alveolar que provoca la migración, la proliferación y la activación de las células del mesénquima, con la formación de focos de fibroblastos y miofibroblastos. Los miofibroblastos activados secretan cantidades exageradas de moléculas de la matriz extracelular con la destrucción posterior de la arquitectura pulmonar.
Asociaciones genéticas
► Factores predisponentes
Numerosos datos contribuyeron al conocimiento de la importancia de los factores genéticos en la fibrosis pulmonar, entre ellas la gran variabilidad de la enfermedad en personas expuestas a concentraciones similares de polvos fibrogénicos, las diferentes respuestas a la fibrosis inducida experimentalmente en ratones y la aparición de fibrosis pulmonar en enfermedades hereditarias con manifestaciones clínicas pleiotrópicas (como la disqueratosis congénita y el síndrome de Hermansky Pudlak).
No obstante, la evidencia más fuerte de la influencia genética en la fibrosis pulmonar proviene de la aparición de la enfermedad en determinadas familias, por ejemplo en gemelos que no fueron criados juntos, en miembros de la familia relacionados genéticamente y en familias multigeneracionales.
► Fibrosis pulmonar familiar
La mayoría de los casos de fibrosis pulmonar idiopática son esporádicos, en personas sin antecedentes familiares, pero en el 5-10% de los casos la enfermedad se produce en dos o más miembros de la misma familia. El patrón de transmisión genética es autosómico dominante. Sin embargo, algunas personas que heredan estos genes mutados nunca sufren la enfermedad, lo que se conoce como penetrancia reducida.
La fibrosis pulmonar familiar y la esporádica son indistinguibles desde el punto de vista clínico e histológico, pero las formas familiares tienden a aparecer más tempranamente y pueden mostrar algunas diferencias en las características radiológicas.
El tabaquismo es un factor de riesgo para la aparición de fibrosis pulmonar familiar, lo que sugiere que los irritantes pulmonares podrían acentuar el riesgo genético y que las interacciones genes - ambiente son esenciales para desencadenar la enfermedad.
Además, puede haber heterogeneidad anatomopatológica entre miembros de la misma familia, que sugiere que diferentes formas de neumonía intersticial idiopática podrían compartir orígenes genéticos comunes y que otros factores genéticos o ambientales actúan sobre éstos para producir diferentes manifestaciones fenotípicas de la enfermedad.
► Mutaciones de la proteína C surfactante
Una de las primeras asociaciones genéticas identificadas fue una mutación en SFTPC, gen que codifica para la proteína C surfactante. Esta proteína es esencial para la función pulmonar y la homeostasis tras el nacimiento.
Posteriormente se identificaron una mutación autosómica dominante en una niña recién nacida y en su madre, que causa la deleción de exon 4 en el gen SFTPC gene y una mutación en SFTPC de sentido erróneo heterozigota (L188Q), que produce el precursor de una proteína mutada que se acumula en el retículo endoplásmico. Su acumulación activa la respuesta de proteína no plegada, una cascada de actividades que, aunque tiene por objeto proteger la célula, podría llevar a la apoptosis de la célula epitelial alveolar en casos de activación prolongada o intensa. Lo mismo sucede en la fibrosis pulmonar idiopática esporádica, lo que sugiere una vía patógena común. También las mutaciones en SFTPA2 parecen predisponer a la fibrosis pulmonar familiar.
► Mutaciones de la telomerasa
Los telómeros son regiones de ADN no codificante, que protegen los extremos de los cromosomas contra la degradación. El complejo telomerasa mantiene la integridad de los telómeros al añadir secuencias a los extremos de los cromosomas. Sus dos componentes principales son la transcriptasa inversa (TERT) y la porción ARN (TERC).
Históricamente, se notificó fibrosis pulmonar en algunos pacientes con disqueratosis congénita, una enfermedad rara que cursa con insuficiencia medular y manifestaciones mucocutáneas. El 20% de los casos sufren también fibrosis pulmonar, que es la segunda causa de muerte en esta enfermedad (la insuficiencia medular es la primera).
Las formas autosómicas dominantes de disqueratosis congénita están ligadas a mutaciones heterocigóticas de TERC y TERT, pero la forma ligada al cromosoma X es causada por mutación de DKC1, que codifica para la disquerina, una proteína asociada a la telomerasa. Aunque la mutación de DKC1 es identificable en un tercio de los pacientes con disqueratosis congénita, la longitud del telómero está reducida en todos los pacientes, lo que sugiere que otros factores genéticos o ambientales contribuyen a la patogenia de la enfermedad. Al igual que en la disqueratosis congénita, gran proporción de pacientes con fibrosis pulmonar familiar tienen mantenimiento anormal de los telómeros.
Se hallaron mutaciones de TERT o TERC en el 10% de pacientes con fibrosis pulmonar hereditaria autosómica dominante y en el 1-3% de pacientes con fibrosis pulmonar idiopática esporádica. La característica histopatológica más frecuente en pacientes con estas mutaciones es la neumonía intersticial, pero también se hallaron características heterogéneas de neumonía intersticial idiopática en individuos con cualquier mutación y en familiares con la misma mutación.
Esto indica que los telómeros cortos pueden causar diversas alteraciones histopatológicas. Son comunes en pacientes con fibrosis pulmonar idiopática que no poseen alelos mutantes para la telomerasa; los pacientes con enfermedad esporádica tienen telómeros mucho más cortos que controles de la misma edad. El acortamiento de los telómeros puede ser adquirido, entre otras causas por el envejecimiento, que podría ser en parte la razón de la mayor frecuencia de la fibrosis pulmonar idiopática en ancianos.
Las disminuciones de la longitud de los telómeros aumentan el riesgo de enfermedad extrapulmonar, como la cirrosis hepática o la diabetes. Ambas son más frecuentes de lo que cabría esperar en la fibrosis pulmonar idiopática.
► Estudio del genoma
Los estudios de asociación de todo el genoma (GWAS por las siglas del inglés) son un método no sesgado y libre de hipótesis basado sobre los datos aportados por el proyecto HapMap. Los GWAS pudieron identificar miles de locus que afectan la sensibilidad a trastornos complejos.
En un barrido de todo el genoma de seis familias de Finlandia, con numerosos miembros afectados con fibrosis pulmonar idiopática, se halló un haplotipo compartido en el cromosoma 4q31 que fue más común en individuos afectados que en controles sanos. Este haplotipo albergaba a ELMOD2, un gen expresado en ls macrófagos alveolares y el epitelio alveolar.
El primer GWAS efectuado para la fibrosis pulmonar idiopática, que examinó más de 200 000 polimorfismos de un solo nucleótido en 159 pacientes japoneses y 934 controles, confirmó la asociación con TERT y la contribución de la replicación y la estabilización de telómeros en la patogenia de la fibrosis pulmonar idiopática. Noth y col efectuaron un GWAS seguido por dos estudios de casos y controles en una gran cohorte de pacientes con fibrosis pulmonar idiopática. Identificaron nuevas variantes genéticas en TOLLIP (11p15.59) y SPPL2C (17q21.31) asociadas con susceptibilidad a la fibrosis pulmonar idiopática.
La importancia de TOLLIP en la regulación del sistema inmunitario lo transforma en un candidato biológicamente verosímil en la patogenia de la fibrosis pulmonar idiopática. Fingerlin y col efectuaron el mayor GWAS hasta ahora en la fibrosis pulmonar idiopática, con 2492 pacientes con neumonía intersticial idiopática (la mayoría de los cuales sufrían fibrosis pulmonar idiopática) y más de 6000 controles.
Confirmaron tres asociaciones genéticas ya conocidas (TERC en 3q26, TERT en 5p15 y MUC5B en 11p15) e identificaron siete nuevos locus de riesgo dentro de los genes que participan en la defensa del huésped, la adhesión intercelular y la reparación del ADN. La asociación entre el riesgo de fibrosis pulmonar idiopática y OBFC1, otro gen que afecta la longitud del telómero, destaca la importancia de la senescencia celular precoz en el desarrollo de la fibrosis pulmonar.
Otro gen ligado al riesgo de fibrosis pulmonar es DSP (6p24.3), que codifica para los desmosomas. Éstos son un componente de las uniones intercelulares adhesivas y son esenciales para conservar la integridad de los tejidos que sufren estrés mecánico, como la periferia pulmonar durante la respiración.
► Genes de mucina
MUC5AC y MUC5B son las principales proteínas formadoras de gel en las secreciones de las vías respiratorias. Seibold y col informaron que el 38% de los pacientes con fibrosis pulmonar idiopática, el 34% de los pacientes con neumonía intersticial familiar y el 9% de controles sanos eran portadores del alelo menor de una variante común ubicada tres pares de kilobases antes del lugar del comienzo de la transcripción MUC5B (rs35705950). Los pacientes con dos copias del alelo mutante MUC5B rs35705950 tenían aproximadamente 20 veces más riesgo de sufrir tanto la enfermedad esporádica como la familiar.
Ser portador del alelo mutante MUC5B se asocia no sólo con el riesgo de sufrir fibrosis pulmonar idiopática, sino también con mejor pronóstico. Hasta ahora, es difícil explicar el motivo de este hallazgo (ie, la misma variante confiere efectos opuestos sobre la susceptibilidad y la supervivencia).
A pesar de la solidez de la asociación de MUC5B con la fibrosis pulmonar idiopática esporádica y familiar, sólo una pequeña proporción de portadores de esta variante en la población general sufrirá a la larga fibrosis pulmonar idiopática, lo que sugiere que la predisposición genética es probablemente necesaria, pero no suficiente, para iniciar las vías patógenas.
La incidencia anual de la FPI está aumentando y se estima de 4,6 a 16,3 por 100.000 personas, con una prevalencia de 13 a 20 casos cada 100 000. La enfermedad predomina más en los hombres (1,5-1,7:1) y su frecuencia aumenta con la edad. Los factores de riesgo ambientales importantes son el tabaquismo y la exposición al polvo de metales y madera. La transmisión genética se produce en alrededor de 0,5-3,7% de los pacientes con FPI, aunque esta frecuencia puede ser mayor en las familias afectadas con un patrón de transmisión vertical autosómico dominante con reducción de la penetrancia. En general, los casos familiares de FPI se pierden. Todavía falta definir el efecto sobre la evolución clínica de la FPI de varias condiciones comórbidas - obesidad, diabetes mellitas, reflujo gastroesofágico, hipertensión pulmonar, apnea obstructiva del sueño, enfermedad arterial coronaria, y enfisema.
"La forma más frecuente y letal de las neumonías intersticiales idiopáticas es la fibrosis pulmonar idiopática"
La fibrogénesis es una parte fisiológica y autolimitada del proceso de cicatrización en respuesta a la lesión tisular. Si la lesión es grave o repetitiva o si el proceso de reparación es descontrolado, la producción exagerada de componentes de la matriz extracelular puede conducir a excesivo crecimiento tisular, fibrosis e insuficiencia orgánica -evolución común de las enfermedades fibróticas que pueden afectar a muchos órganos, entre ellos los pulmones. Algunas formas de fibrosis pulmonar se asocian con exposiciones ambientales u ocupacionales, como la asbestosis o la inhalación de aerosol de antígenos orgánicos (eg, Saccharopolyspora rectivirgula), con enfermedades reumatológicas (eg, esclerosis sistémica o artritis reumatoide), efectos tóxicos de fármacos (eg, amiodarona), o exposición a radiaciones.
Las neumonías intersticiales idiopáticas son de causa desconocida y probablemente generadas por la interacción entre una o más predisposiciones genéticas y desencadenantes ambientales, como el humo de cigarrillo y consisten en un un grupo diverso caracterizado por diversas formas de inflamación y fibrosis como resultado de daño del parénquima pulmonar, pero que comparten características clínicas, radiológicas y fisiológicas similares.
La forma más frecuente y letal de las neumonías intersticiales idiopáticas es la fibrosis pulmonar idiopática, una enfermedad devastadora que afecta principalmente a adultos mayores (mediana de edad al diagnóstico 66 años) con una mediana de supervivencia de sólo 2-3 años, mucho menor que la supervivencia de pacientes con otras neumonías intersticiales idiopáticas crónicas. Actualmente no hay tratamientos para la fibrosis pulmonar idiopática, salvo el trasplante pulmonar, aunque algunos fármacos (eg, pirfenidona y nintedanib) disminuyen la velocidad del deterioro funcional.
► Desde la enfermedad inflamatoria hasta la enfermedad por alteración epitelial
La inflamación tiene un papel fundamental en la mayoría de las enfermedades intersticiales del pulmón y, si es crónica, evoluciona a la fibrosis. Sin embargo, con la redefinición de FPI como una enfermedad distinta caracterizada por un patrón típico de NIU, la reacción fibrótica progresiva de la FPI se asocia con un proceso activado por los fibroblastos dependientes del epitelio y una escasa respuesta al tratamiento con antiinflamatorios. Sin embargo, en un subgrupo de pacientes, la desregulación de los mecanismos inmunes adaptativos y la inflamación posterior podrían representar un papel en el comienzo o la progresión de la enfermedad. Por lo tanto, la fibrosis pulmonar epitelial podría tener al menos dos rutas celulares diferentes - la vía inflamatoria y la vía epitelial, las que podrían dar lugar a la fibrosis pulmonar.
► Lesiones epiteliales y activación: interacciones genéticas y ambientales
Varios factores ambientales pueden contribuir a las lesiones epiteliales y la apoptosis, como el tabaquismo y la microaspiración crónica silenciosa. Por otra parte, la infección viral crónica, principalmente por el virus herpes, podría contribuir a la patogénesis de la FPI.
No hay factores genéticos asociados consistentemente con la FPI esporádica. En algunos casos familiares de fibrosis pulmonar que tienen mutaciones en la proteína surfactante C aparecen alteraciones en la respuesta de la proteína no plegada, una proteína hidrofóbica expresada exclusivamente por células alveolares epiteliales activadas tipo II (CEA II), activadas en forma aberrante. El cambio de sentido o las mutaciones en la deleción corta de esta proteína provocan la producción de proteínas mal plegadas, que, al acumularse o agruparse en complejos pueden causar la lesión de las células epiteliales. Un polimorfismo común en la región promotora del gen de la mucina 5B (MUC5B) se asocia con la neumonía intersticial familiar y la FP esporádica.
El estudio del genoma de 6 familias con FPI familiar mostró un haplotipo compartido en el cromosoma 4q31, que fue significativamente más frecuente en los pacientes que en los controles de la población. Este haplotipo albergaba ELMOD2, un gen expresado en el pulmón, pero cuya expresión es significativamente menor en el pulmón de la FPI, comparado con la de los pulmones sanos. El ELMOD2 es esencial para los procesos celulares y podría tener un efecto antiviral en las CEA. También se han identificado mutaciones de la telomerasa.
A pesar de la lesión epitelial y la apoptosis, hay un aumento del número de neumocitos tipo II hiperplásicos e hipertróficos, una característica notable de los pulmones afectados por la FPI. Por otra parte, se observan células epiteliales grandes y alargadas o atenuados. También se ha informado la presencia de epitelio tipo bronquiolar y de metaplasia escamosa revistiendo las lesiones en panal de abejas. Las células epiteliales son muy activas, lo que lleva a la desregulación del proceso de reparación que parece estar perpetuamente activado, incluso en ausencia del estímulo primario.
Nuevas pruebas indican que la desregulación de algunas vías embriológicas podría explicar el comportamiento anormal del CEA y tal vez de los fibroblastos. Los Ligandos Wnt forman una gran familia que secreta glucoproteínas esenciales para los procesos morfogenéticos. Los resultados de varios estudios indican que el epitelio alveolar y los fibroblastos expresan en exceso a miembros de la vía Wnt/Wg en los pulmones de la FPI. Por otra parte, hay una gran acumulación nuclear de catenina β en las CEA y los fibroblastos, lo que sugiere que en ambos tipos celulares la vía Wnt-β-catenina está activada.
El gen homólogo de la fosfatasa y la tensina (PTEN) es crucial para el desarrollo. En los adultos, participa en la regulación de los procesos fisiológicos tales como la polaridad, la proliferación, y la apoptosis de las células. En los pacientes con FPI, la expresión de PTEN está regulada hacia abajo en los miofibroblastos dentro de los focos fibroblástcios, lo que podría explicar su supuesta resistencia a la apoptosis. La interacción de la integrina ß1-colágeno en los fibroblastos normales activa al PTEN, que es también un regulador negativo del crecimiento, mientras que el mecanismo de retroalimentación negativo es defectuoso en los fibroblastos de la FPI.
El gen sonic hedgehog (Shh) es un morfogen esencial para la definición de los patrones durante la embriogénesis. Este ligando del desarrollo permite que las células evadan la apoptosis y la detención del ciclo celular, favoreciendo la proliferación. En los pulmones afectados por la FPI hay una fuerte expresión de Shh, principalmente en las células epiteliales que recubren los quistes de la lesión en panal de abejas.
Las proteínas morfogenéticas óseas pertenecen a la superfamilia del factor de transformación del crecimiento β (TGF β) y tienen un papel esencial en el estado embrionario y el desarrollo posnatal. En los adultos, la reactivación de la expresión de los antagonistas de la proteína morfogenética ósea puede contribuir a la progresión de algunas enfermedades crónicas degenerativas.
Se ha informado que en los fibroblastos de los pulmones de la FPI existe un aumento de la expresión de gremlina, un fuente antagonista de una proteína morfogenética ósea. El aumento de las concentraciones de gremlina podría atenuar la fosforilación mediada por la señalización de la proteína morfogenética ósea en los pulmones, dando lugar a la transición epitelio-mesénquima inducida por el TGFβ1 y a la disminución de la apoptosis de los miofibroblastos.
► Efectos profibróticos de las CEA en forma aberrante en el microambiente pulmonar
Un proceso patológico importante en la FPI es la activación de la cascada de la coagulación, que tiene varios efectos profibróticos. En la FPI, el complejo de factores tisulares-factor VIIa-Factor X se forma en el epitelio alveolar, permitiendo la activación del factor X que a su vez estimula los fibroblastos en las regiones fibróticas subyacentes. La matriz provisional, formada por fibrina y fibronectina, podría estimular la transición epitelio-mesénquima, incluso en ausencia de TGFβ1. Por otra parte, la trombina y el factor X activados inducen la diferenciación de los fibroblastos pulmonares a miofibroblastos a través de la vía del receptor 1 activado por la proteasa. Estos hallazgos proporcionan pruebas convincentes de que en la FPI se activa la señalización procoagulante y que la función de la fibrinólisis alveolar deficiente, causada principalmente por las células epiteliales, representa un papel importante en la respuesta fibrótica del pulmón.
En los tejidos dañados, los fibroblastos se activan y diferencian en miofibroblastos, los cuales son células contráctiles especializadas con mayor potencial profibrótico que los fibroblastos. En los focos fibroblásticos, estas células provocan el depósito exagerado de matriz extracelular, que es el sello distintivo del proceso de cicatrización que conduce a la destrucción de la arquitectura pulmonar. Se desconoce el origen de los fibroblastos y los miofibroblastos y las razones por las cuales en la FPI se organizan en focos morfológicamente distintos.
Hay muchas pruebas de que las CEA son la fuente principal de los mediadores que funcionan como factores quimiotácticos o mitógenos de las células del mesénquima, como el factor de crecimiento derivado de las plaquetas, el factor de necrosis tumoral α (TGF α) y la endotelina 1. Estos factores probablemente contribuyen sobre todo a la migración, la proliferación y la diferenciación de las células del mesénquima.
Hay un influjo de circulación de fibrocitos en los pulmones de la FPI, los que constituyen una subpoblación particular de leucocitos caracterizados porque expresan marcadores de células hematopoyéticas (CD45, CD34) y mesenquimales (colágeno I, fibronectina). La mayoría de los fibrocitos circulantes expresan el receptor de quimiocinas CXCR4, lo que sugiere que el eje CXCR4-CXCL12 es crucial para el traslado en los pulmones de la FPI. Las CEA de estos pacientes expresan gran cantidad de CXCL12, lo que probablemente constituye el gradiente quimiotáctico necesario para el tráfico de fibrocitos positivos para CXCR4.
El epitelio podría contribuir directamente a la expansión de la población de fibroblastos y miofibroblastos a través de la transición epitelio-mesénquima. En este proceso, las células epiteliales adquieren propiedades mesenquimáticas por las cuales aumentan su capacidad para moverse y sintetizar matriz intersticial. En la FPI, las células mesenquimáticas locales, los fibrocitos circulantes y la transición epitelio-mesénquima participan en la expansión de los fibroblastos y los miofibroblastos. Sin embargo, no se sabe en qué medida participa cada uno en el inicio o la perpetuación de la enfermedad.
► Diferenciación de los fibroblastos en miofibroblastos
En esta diferenciación intervienen 3 factores principales: la tensión mecánica (que induce la diferenciación de los protomiofibroblastos), el aumento local de la actividad del TGFβ1 y la presencia de proteínas especializadas de la matriz. Los miofibroblastos causan una acumulación adicional exagerada de matriz celular (fibrosis) y contribuyen a la interrupción de la membrana basal y la muerte de las células epiteliales.
La eliminación de los miofibroblastos por la apoptosis es esencial durante el proceso normal de cicatrización, que no parece producirse en los focos fibroblásticos de la FPI. Las señales del microambiente como el TGFβ1 y la endotelina 1 (en su mayoría sintetizados por las CEA en la FPI) podrían promover la resistencia a la apoptosis de los fibroblastos a través de las vías de señalización que involucran PI3K/AKT. Sin embargo, no hay evidencia convincente de que la reducción de la susceptibilidad a la apoptosis ocasiones la persistencia de los miofibroblastos in vivo. Por otra parte, se desconocen las razones por las cuales aparentemente los fibroblastos y los miofibroblastos sobreviven mientras que las células epiteliales mueren dentro del mismo microentorno. Una explicación posible sería la deficiencia de prostaglandina E2 que se observa en la FPI, que aumenta la sensibilidad de las CEA a la apoptosis pero la disminuye en los fibroblastos.
► Ausencia de neumocitos tipo I
Las CEA tipo I cubren más del 90% de la superficie alveolar en la periferia del pulmón, y la interconexión con los capilares pulmonares proporciona una superficie fácilmente permeable para los gases. Los pacientes con FPI tienen una importante pérdida de neumocitos tipo I, aunque el efecto putativo de este proceso patológico en la respuesta fibrótica no está claro. Por otra parte, la transdiferenciación de los neumocitos tipo II en neumocitos tipo I, imprescindible para volver a establecer un epitelio alveolar funcional, está profundamente alterada en la FPI, debido a las anormalidades severas de la matriz extracelular y la membrana basal del epitelio interrumpida.
La pérdida de las CEA tipo I podría provocar la reducción de algunas moléculas antifibróticas importantes (por ej. la caveolina-1). Sin embargo, no se sabe si en la patogénesis de la FPI está involucrada la disminución de las concentraciones de caveolina-1, específicamente atribuible a la pérdida de los neumocitos tipo I. Por otra parte, el receptor de los productos finales de la glicación avanzada (RPFGA) también está disminuido en la FPI. El RPFGA es un miembro de la superfamilia de inmunoglobulinas de los receptores de la superficie celular que son altamente expresados por las CEA tipo I normales. La pérdida de este receptor podría resultar en una disminución de la unión de las CEA tipo I a la membrana basal, lo que impide la correcta reepitelización de los alvéolos durante la fibrogénesis.
► Metaloproteinasas de la matriz y remodelación pulmonar anormal
Entre los genes mayormente expresados en la FPI están las metaloproteinasas 7 (MMP7), MMP1 y MMP2 pero también se han reportado otras MMP en el líquido del lavado broncoalveolar (MMP3, MMP8 y MMP9) o se han localizado mediante técnicas de inmunohistoquímica (por ej., MMP de membrana). La mayoría de estas MMP se localizan en las CEA, pero pocas están en los focos fibroblásticos. La MMP1 se localiza principalmente en el epitelio y está casi ausente en el compartimiento intersticial, donde se acumula el colágeno. La MMP7 y la osteopontina interactúan para afectar el fenotipo de la FPI (por ej., MMP7 es inducida por la osteopontina y la osteopontina también se escinde y es activada por la MMP7).
Tanto la MMP1 como la MMP7 facilitan la migración de las células epiteliales, reduciendo la afinidad de la integrina α2β1. El aumento de la expresión de MMP1 podría estar en parte asociado a dos polimorfismos del gen que afecta la capacidad de respuesta a la transcripción. Las concentraciones de MMP2 están aumentadas tanto en las CEA alveolares como bronquiolares y en los focos fibroblásticos. Los principales activadores de la proMMP2 son miembros de la familia MMP de la membrana que también se expresa fuertemente en los pulmones de la FPI. Por lo tanto, la mayor cantidad de MMP2 activa podría ser atribuida a la acción de estas enzimas. La MMP9 también está elevada en la FPI, y un aumento de su forma activa en el lavado broncoalveolar se asocia con un fenotipo clínico acelerado.
► Angiogénesis y remodelación vascular
La neovascularización es un proceso fundamental en el tejido de reparación después de una lesión y depende del equilibrio entre diversos factores, principalmente las quimiocinas que promueven o inhiben la angiogénesis. No se conoce cuál es el papel de la angiogénesis el pulmón de la fibrosis pulmonar. En los pulmones se produce una remodelación vascular aberrante, pero las áreas fibróticas tienen menos vasos sanguíneos mientras que el tejido de las zonas no fibróticas está muy vascularizado. Casi no hay capilares dentro de los focos fibroblásticos, lo que indica que el proceso fibrótico en la FPI no necesita la neovascularización. Por lo tanto, en ciertas configuraciones patológicas, el aumento de la angiogénesis parece tener un papel fibrogénico mientras que en otras, la angiogénesis favorece la fibrosis.
► Procesos que vinculan el envejecimiento con la FPI
Los mecanismos que vinculan el envejecimiento con la FPI se desconocen. En general, la TC de alta resolución muestra signos en los individuos asintomáticos mayores (≥75 años) pero no en los más jóvenes. Un posible mecanismo está relacionado con un acortamiento acelerado de los telómeros, los que en cada división celular se acortan al alcanzar una longitud crítica, luego activan un sitio dependiente de p53 que conduce a la apoptosis o senescencia replicativa. Se cree que los telómeros cortos comprometen la replicación de las células progenitoras que se quedan en los tejidos después de la lesión. Las personas podrían estar en mayor riesgo de desarrollar FPI. Algunos pacientes con otras formas de FPI también tienen telómeros acortados, aumentando la posibilidad de mecanismos compartidos.
Sin embargo, el acortamiento de los telómeros también se observa en la enfermedad pulmonar obstructiva crónica, otra enfermedad relacionada con la edad pero completamente diferente. Por otra parte, se ha observado que los ratones con deficiencia de telomerasa que tienen un acortamiento secuencial espontáneo de los telómeros desarrollan lesiones enfisematosas.
Las CEA de esos ratones activan la vía de la respuesta al estrés y la apaptosis espontánea, indicando que el acortamiento de los telómeros provoca daño de las células epiteliales pero la consecuencia es el enfisema y no la fibrosis. Adicionalmente, en el enfisema humano también se ha observado apoptosis epitelial alveolar. No obstante, el cambio en la población total en la distribución de los telómeros sugiere que el acortamiento de los telómeros puede ser un cofactor patógeno para la FPI.
El envejecimiento también se asocia con el aumento del estrés oxidativo como resultado del desequilibrio entre los prooxidantes (especies de O2 reactivo y nitrógeno) y antioxidantes (por ej., superóxido dismutasa, glutatión). Las consecuencias son el daño directo del ADN, la oxidación de los ácidos grasos poliinsaturados en las membranas celulares y la inactivación de las enzimas. El estrés oxidativo excesivo tiene varios efectos nocivos que pueden contribuir a la patogénesis de la FPI, incluyendo la activación de vías de la vía de señalización redox-sensible, los cambios en la expresión de citocinas o quimiocinas, la modificación del balance las proteasas o antiproteasas, la inducción de la apoptosis y la activación de los fibroblastos.
Los procesos epigenéticos incluyen alteraciones transmisibles en la expresión genética causadas por otros mecanismos diferentes de los cambios en la secuencia del ADN, y estos procesos son esenciales para el normal desarrollo y mantenimiento de los patrones de expresión de los genes titulares específicos. Los mecanismos epigenéticos más comunes son la metilación del ADN, las modificaciones postraducción de las histonas y los efectos de transcripción de las moléculas no codificantes del ARN, como la microRNA.
En los ancianos (>65 años) hay una pérdida progresiva de la metilación del ADN en elementos repetitivos dispersos de todo el genoma que parece ser proporcional a la expectativa de vida. En el inicio y la progresión del cáncer interviene una reprogramación aberrante del epigenoma. Los cambios epigenéticos contribuyen al comportamiento agresivo de los fibroblastos en la FPI. Thy1, un receptor que inhibe la diferenciación de los fibroblastos en miofibroblastos y que disminuye la actividad fibrogénica, no se expresa en los focos fibroblásticos in vivo.
La pérdida de este receptor ocurre por el silenciamiento epigenético secundario a la hipermetilación de la islas de citosina-guanina en el promotor del gen. En comparación con los fibroblastos normales, los fibroblastos de la FPI expresan menos ciclooxigenasa 2 y sintetizan menos prostaglandina E2, un regulador hacia abajo potente de la activación de los fibroblastos. La menor expresión de ciclooxigenasa-2 en los fibroblastos de la FPI parece estar causada por anormalidades epigenéticas de la acetilación de las histonas que inhibe los factores de transcripción activados de la unión al promotor 2 de la ciclooxigenasa.
Los microARN son reguladores postranscripción que se unen a secuencias específicas, bloqueando la traducción o causando la degradación del ARN mensajero diana, lo que resulta en el silenciamiento de los genes. Las microARN probablemente controlan a la mayoría de las rutas y redes biológicas. Durante el envejecimiento, la desregulación de la expresión de las microRNA se produce generalmente en grupos, lo que indica que las acciones podrían estar funcionalmente coordinadas juntas por los reguladores de transcripción comunes.
La disrupción de estas alianzas vitales dependientes de la edad podría contribuir al desarrollo de las enfermedades en las personas de edad avanzada. Como ejemplo, la regulación negativa de algunas micro ARN (miR1, mirR30), junto con la regulación hacia arriba de otras (miR20a, miR21) son frecuentes en los pacientes con hipertrofia cardiaca y arteriosclerosis coronaria. Las microARN se expresan en la FPI en forma diferente que en los pulmones normales, con una disminución significativa de microARN let-7d. La inhibición de let-7d in vitro indujo la transición epitelio-mesénquima mientras que la inhibición in vivo causó fibrosis alveolar septal. Del mismo modo, en la FPI, los pulmones tienen una regulación hacia arriba de la miR21 y una regulación hacia abajo de la miR29, principalmente localizadas en los miofibroblastos.
El aumento de los niveles de miR21 promovió la actividad fibrogénica de TGFβ1 en los fibroblastos, mientras que la regulación hacia debajo de miR-21 atenuó esta actividad. Por el contrario, los niveles de miR29 se correlacionan inversamente con la expresión de varios genes profibróticos diana y con la gravedad de la fibrosis. Por lo tanto, la desregulación epigenética que influye en el fenotipo del envejecimiento podría aumentar el riesgo de desarrollar FPI si se alcanza un umbral crucial de epimutaciones.
Fibrosis pulmonar asociada con enfermedades reumatológicas
► Diferenciación de la fibrosis pulmonar según los mecanismos patógenos
El pulmón sería un órgano importante en el desarrollo de enfermedades autoinmunes porque podría sensibilizar a las células T patógenas antes de ser reclutadas a los órganos vulnerables en los que atacan los autoantígenos y desencadenan la autoinmunidad. Asimismo, cada vez hay más conciencia de la afectación pulmonar (especialmente fibrosis) en las enfermedades del tejido conjuntivo.
Diferenciar la fibrosis pulmonar asociada con estas enfermedades de la fibrosis pulmonar idiopática puede ser difícil porque la afectación pulmonar intersticial puede ser la manifestación inicial en las enfermedades del tejido conjuntivo. Además, se observa neumonía intersticial, histológica o radiológica, en estas enfermedades, en especial en la artritis reumatoide.
Al evaluar a pacientes jóvenes con enfermedad pulmonar intersticial, especialmente a mujeres, es necesario un alto índice de sospecha de una enfermedad subyacente del tejido conjuntivo, ya que la fibrosis pulmonar idiopática es rara antes de los 50 años.
► Artritis reumatoide
La artritis reumatoide se produce como resultado de la exposición ambiental en personas predispuestas genéticamente. Los factores de riesgo en un subgrupo de pacientes con esta enfermedad en los que se identificaron anticuerpos anti-proteínas citrulinadas (ACPA) incluyen ciertos alelos HLA (llamados epítopos compartidos) y la exposición al humo de cigarrillo.
La fibrosis pulmonar es una complicación frecuente de la artritis reumatoide. El 4-68% de los pacientes tienen algún tipo de alteración pulmonar y en un estudio el 10-12% de los pacientes tenían fibrosis difusa. La enfermedad pulmonar intersticial podría aparecer años antes de las manifestaciones extrapulmonares y un estudio mostró que era la primera manifestación en el 3,5% de los pacientes con enfermedad del tejido conjuntivo.
Hasta ahora se han identificado pocas asociaciones genéticas entre la artritis reumatoide y la fibrosis pulmonar; en la mayoría participan los locus del gen MHC en el cromosoma 6, cuya función principal es la presentación de antígenos a las células T al inicio de una respuesta inmunitaria adaptativa.
Las alteraciones del parénquima pulmonar en la TC de alta resolución al inicio de la enfermedad en los pacientes con ACPA y artritis reumatoide sugestivas de fibrosis pulmonar son más frecuentes que en los pacientes con artritis reumatoide sin ACPA y los controles. Estas diferencias son independientes del tabaquismo y sugieren una asociación entre la predisposición genética a desarrollar anticuerpos ACPA y la fibrosis pulmonar. Algunos datos sugieren que estos anticuerpos se producen en el pulmón.
► Esclerosis sistémica (Esclerodermia)
La fibrosis pulmonar es una causa importante de morbimortalidad en pacientes con esclerosis sistémica y el grado de afectación fibrótica del pulmón es un factor pronóstico significativo de la evolución. La identificación temprana de los pacientes cuya fibrosis pulmonar tiene más probabilidades de progresar es muy importante. Los anticuerpos anti-topoisomerasa (ATA; también llamados anticuerpos antiscleroderma 70) están muy asociados con la aparición de fibrosis pulmonar en la esclerosis sistémica.
Los ATA se vinculan con los alelos MHC, especialmente HLA-DRB1*11, HLA-DPB1*1301 y HLADRB1*15. Estas asociaciones sugieren que factores genéticos afectan la producción de autoanticuerpos, que a su vez está muy ligada con la presencia de fibrosis pulmonar en la esclerosis sistémica.
Los investigadores que efectuaron el primer estudio GWAS en la esclerosis sistémica identificaron tres locus no HLA asociados con la enfermedad, entre ellos el factor 5 regulador de interferón. Los mismos autores mostraron una asociación entre la presencia del polimorfismo de un solo nucleótido rs4728142 en la región promotora de IRF5 y las siguientes características en pacientes: afectación pulmonar intersticial más leve, mayor conservación de la función pulmonar y supervivencia más prolongada.
Este efecto protector de la variante IRF5 rs4728142 coincide también con el patrón de activación del interferón tipo 1 en la esclerosis sistémica, como asimismo con el resultado de los intentos de tratar a los pacientes con esclerosis sistémica con interferón-α, que producen el empeoramiento de la enfermedad pulmonar.
Variantes de NLRP1 también se asociaron con la producción de ATA y la fibrosis pulmonar, lo que sugiere la importancia de la inmunidad innata en la aparición de fibrosis pulmonar en pacientes con esclerosis sistémica.
► Sarcoidosis
La sarcoidosis es un trastorno granulomatoso sistémico de causa desconocida, con afectación pulmonar casi invariable. La fibrosis pulmonar afecta hasta al 25% de los pacientes. Estudios epidemiológicos que muestran los efectos para ciertos orígenes étnicos y un riesgo aproximadamente cinco veces mayor de aparición de sarcoidosis entre los hermanos del paciente, sugieren la participación genética en el riesgo de sufrir sarcoidosis.
Las investigaciones indicaron hasta 80 veces mayor riesgo para hermanos gemelos de pacientes con sarcoidosis. A pesar de este riesgo, pocos hermanos de pacientes con sarcoidosis (<1%) sufren la enfermedad, lo que sugiere la importancia de los factores ambientales.
Los genes HLA clase II tienen fuerte participación en la susceptibilidad a la sarcoidosis. En general, HLA-DRB1*01 y HLADRB1* 04 protegen contra la enfermedad, mientras HLA-DRB1*03, HLA-DRB1*11, HLA-DRB1*12, HLADRB1* 4, y HLA-DRB1*15 se asocian con aumento del riesgo.
Los pacientes con la forma crónica de sarcoidosis tienen mayor riesgo de fibrosis pulmonar. De hecho, las granulomas característicos de la sarcoidosis se resuelven espontáneamente o de lo contrario progresan a la fibrosis. HLA-DRB1*03 se asocia fuertemente con el curso favorable de la enfermedad y los pacientes portadores de HLA-DRB1*03 muy raras veces sufren fibrosis pulmonar.
Entre los pacientes con signos de síndrome de Lofgren- comienzo agudo de artritis bilateral de tobillos, eritema nudoso, adenopatía hiliar bilateral y fiebre- casi todos los portadores de HLA-DRB1*03 se recuperan dentro de los 2 años del comienzo de la enfermedad. En cambio, sólo la mitad de los pacientes con síndrome de Lofgren sin HLA-DRB1*03 se recuperan en ese tiempo. HLA-DRB1*15 se asoció repetidas veces con enfermedad crónica y mayor riesgo de fibrosis pulmonar.
Las investigaciones indicaron asimismo una asociación entre ser portador de una variante no funcional dentro de BTNL2 y aumento del riesgo de fibrosis pulmonar. Variantes genéticas de GREM1, que codifica la gremlina para mantener la homeostasis tisular y la regeneración tras una lesión, se asocian con fibrosis en pacientes con sarcoidosis.
► ¿Qué podrían significar los hallazgos genéticos para la patogenia de la fibrosis pulmonar idiopática?
Un conjunto creciente de datos sugiere que la fibrosis pulmonar está determinada genéticamente. La pregunta es cómo se pueden incorporar las alteraciones genéticas identificadas por los investigadores a un modelo patogénico unificado para la fibrosis pulmonar idiopática.
Las mutaciones asociadas con la enfermedad en SFTPC y SFTPA2, que son expresadas exclusivamente por las células epiteliales alveolares tipo 2, señalan hacia la lesión de las células epiteliales y la pérdida de su capacidad de reparación, mediada por la acumulación proteínas con trastornos de su plegamiento en el retículo endoplásmico y cambios en la respuesta de las proteínas no plegadas.
La deficiencia de la proteína C surfactante también podría llevar a la fibrosis pulmonar. Además, los cambios en los genes específicos de las células epiteliales alveolares podrían explicar, al menos parcialmente, porqué la fibrosis en la fibrosis pulmonar idiopática se produce sólo en el pulmón.
Al contrario de SFTPC y SFTPA2, los genes de la telomerasa (eg, TERT y TERC) también son expresados por células extrapulmonares, especialmente las células madre, y plantean la posibilidad de que los cambios en la renovación y la reparación de las células epiteliales alveolares tras la lesión predispongan a la iniciación y la progresión de la enfermedad.
No obstante, en modelos murinos con fibrosis pulmonar, los telómeros cortos solos no son suficientes para causar enfermedad. La necesidad de repetidas lesiones también explicaría porqué la fibrosis pulmonar idiopática se manifiesta tardíamente en la vida.
MUC5B es expresada por las células epiteliales bronquiolares (no las alveolares) y el alelo de riesgo MUC5B se asocia con mayor expresión de proteínas que en personas sanas-limitado a las zonas fibróticas del pulmón. Debido a que las mucinas son importantes para la defensa mucosa, un MUC5B excesivo (eg, exceso de producción de moco) podría causar lesión pulmonar al hacer más lenta la depuración pulmonar de las toxinas y los microrganismos inhalados.
El MUC5B excesivo también podría dificultar la reparación alveolar al interferir con las interacciones de las células epiteliales alveolares con la matriz extracelular o debido a cambios en las propiedades del surfactante y promoción del colapso alveolar.
Hay datos que indican que los quistes en panal de abejas, que constituyen la etapa final irreversible de la fibrosis pulmonar, se originan a partir de las vías aéreas distales, ubicación principal de las células que expresan MUC5B.
Otra hipótesis para vincular la disfunción de la secreción de las vías aéreas y las células alveolares es la de la respuesta de proteínas no plegadas, proceso que puede ser iniciado por muchos estímulos, entre ellos el aumento de la síntesis de mucina. Ambas hipótesis no son mutuamente excluyentes.
En conjunto, estos datos sugieren que en pacientes con fibrosis pulmonar idiopática, la combinación de factores relacionados con la edad, genéticos y ambientales contribuye a que el epitelio alveolar sea especialmente susceptible a una gama de agresiones crónicas endógenas (eg, microaspiración asintomática) o exógenas (eg, agentes infecciosos o tabaquismo).
Las células epiteliales alveolares tipo 2 lesionadas, para restablecer la integridad funcional, liberan citocinas y factores de crecimiento que favorecen el reclutamiento aberrante y la activación de fibroblastos resistentes a la apoptosis, los mediadores clave de la restructuración del tejido fibrótico. A su vez, los fibroblastos forman pequeños conglomerados (focos fibroblásticos) y se diferencian a miofibroblastos, que tienen un fenotipo profibrótico más agresivo. El resultado es la producción exagerada de componentes de la matriz extracelular, que genera restructuración tisular progresiva y cicatrización (con pérdida inevitable de la función).
► ¿Qué podrían significar los datos genéticos para la fibrosis en pacientes que NO sufren fibrosis pulmonar idiopática?
En las enfermedades del tejido conjuntivo y la sarcoidosis, se puede producir fibrosis pulmonar tras una respuesta inmunitaria exagerada e inflamación, pero varios aspectos difieren de la fibrosis pulmonar idiopática. Los pacientes con sarcoidosis habitualmente son más jóvenes, la fibrosis en ellos no es inevitable y en general progresa más lentamente que en pacientes con fibrosis pulmonar idiopática.
La fibrosis en la esclerosis sistémica (esclerodermia) tiene mejor pronóstico y características histopatológicas diferentes que en la fibrosis pulmonar idiopática. Una de las diferencias genéticas es la fuerte asociación entre un polimorfismo (rs35705950) en dirección ascendente de MUCB5 y la fibrosis pulmonar idiopática tanto esporádica como familiar, pero no con la fibrosis en la sarcoidosis o la esclerosis sistémica.
Sin embargo, variantes genéticas de GREM1 se asocian con fibrosis en la sarcoidosis, y la gremlina podría también ser importante en la fibrosis pulmonar idiopática porque está muy sobrexpresada en los pulmones de estos pacientes. Estos datos sugieren que hay muchas rutas diferentes que llevan a la fibrosis en la fibrosis pulmonar idiopática y en las enfermedades del tejido conjuntivo, pero también podrían existir vías comunes.
En las enfermedades reumatológicas, si bien se sabe la existencia de efectos genéticos sobre la producción de autoanticuerpos, no se conoce la relación entre ellos y la fibrosis pulmonar. En la artritis reumatoide, hay una fuerte asociación entre ls epítopos compartidos de HLA y ACPA, y puesto que los pacientes con ACPA parecen tener mayor riesgo de fibrosis pulmonar, hay una asociación indirecta entre estos epítopos y la fibrosis pulmonar. Existe una situación similar en la esclerosis sistémica, en la que ATA se asocia con alelos específicos HLA-DP y HLA-DR y los pacientes con ATA tienen mayor riesgo de sufrir fibrosis pulmonar.
En la sarcoidosis, los genes HLA clase II tienen fuerte participación en la susceptibilidad a la sarcoidosis y también dirigen la enfermedad hacia diferentes fenotipos clínicos. Las fuertes asociaciones con alelos presentadores de antígenos coindicen con la noción de la sarcoidosis como una enfermedad iniciada y perpetuada por la presentación continua de antígenos.
Asociaciones entre alelos MHC y fibrosis pulmonar sugieren que repuestas inmunitarias específicas contra blancos pulmonares desconocidos podrían contribuir a la aparición de fibrosis en las enfermedades inflamatorias sistémicas y ayudar a la tipificación de subconjuntos de pacientes.
► ¿Qué podrían significar los datos genéticos para las opciones terapéuticas o las estrategias para estudios clínicos?
No se dispone de específicos contra el proceso fibrogenético aberrante o contra las repetidas perturbaciones de las células alveolares y que puedan detener la progresión inexorable de la fibrosis pulmonar idiopática.
Sin embargo, las asociaciones genéticas identificadas hasta ahora ampliaron los conocimientos sobre la patogenia de esta enfermedad y sugieren que los investigadores deben prestar más atención al estrés de las células epiteliales alveolares, la defensa del huésped, la adhesión intercelular y la longitud de los telómeros para crear tratamientos eficaces contra la fibrosis pulmonar idiopática.
Los resultados del estudio PANTHER-IPF, que investiga la eficacia del antioxidante mucolítico N-acetilcisteína en la fibrosis pulmonar idiopática, podrían proporcionar más conocimientos sobre los resultados obtenidos al centrarse sobre las vías de los oxidantes que lesionan las células epiteliales.
La combinación de fármacos pleiotrópicos que tratan más de una vía o resultado de los efectos de genes mutantes probablemente surgirá como el método más eficaz para detener la progresión de la enfermedad.
La heterogeneidad de la población de pacientes es un obstáculo importante al diseñar estudios clínicos sobre la fibrosis pulmonar idiopática porque la velocidad de progresión de la enfermedad no es uniforme. Esta variación podría explicar parcialmente la alta tasa de fracasos y la respuesta poco uniforme a los fármacos probados en los estudios clínicos.
Los modelos disponibles de pronóstico de la progresión de la fibrosis pulmonar idiopática se basan sobre parámetros clínicos y fisiológicos, pero aún no están totalmente validados. En 2013, Herazo-Maya y col identificaron varios perfiles de expresión de genes en células mononucleares de sangre periférica que pronosticaban mala evolución en pacientes con fibrosis pulmonar idiopática. Yang y col informaron que la expresión de genes asociados a las cilias define dos fenotipos moleculares únicos de pacientes con fibrosis pulmonar idiopática.
La identificación de características genéticas para pronosticar el comportamiento de la enfermedad y la respuesta terapéutica permitiría adaptar precozmente el tratamiento a cada paciente (o derivarlo para trasplante pulmonar) y recetar los fármacos apropiados.
La FPI pertenece a un gran grupo de más de 200 enfermedades pulmonares llamadas neumopatías intersticiales (NI) que se caracterizan por afectar al intersticio pulmonar,2 el tejido entre los alveolos pulmonares.La FPI es una enfermedad diferenciada entre las neumonías intersticiales idiopáticas (NII), las cuales a su vez son un tipo de NI, también conocidas como neumopatías parenquimatosas difusas (NPD).
La clasificación de 2002 de la American Thoracic Society/European Respiratory Society (ATS/ERS) para las NII se actualizó en 2013.2 En esta nueva clasificación existen tres categorías principales de NII: NII principal, NII raras y NII inclasificables. Las NII principales se agrupan en NI fibrosantes crónicas, que incluye la FPI y la neumonía intersticial no específica [NINE]), las NI relacionadas con el tabaquismo (por ejemplo, la neumopatía intersticial-bronquiolitis [NI-B] y la neumonía intersticial descamativa [NID]; y las NI agudas/subagudas (por ejemplo, neumonía organizante criptogénica [NOC] y neumonía intersticial aguda [NIA].2
El diagnóstico de las NII requiere la exclusión de causas conocidas asociadas a las NI. Se pueden citar como ejemplo de NI de origen conocido la neumonitis por hipersensibilidad, histiocitosis pulmonar de la célula de Langerhan, asbestosis y vasculopatías del colágeno.Sin embargo, estas enfermedades con frecuencia no solo afectan al intersticio, sino también al espacio alveolar, las vías respiratorias periféricas y los vasos sanguíneos.2
La siguiente imagen muestra la nueva clasificación de las NII
Modificada a partir del Consenso Internacional Multidisciplinar de la American Thoracic Society/European Respiratory Society (2013). Am Respir Crit Care Med. 188 (6): 733–748.  EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG |
El médico llevará a cabo un examen físico y hará preguntas acerca de su historia clínica. Igualmente, preguntará si usted ha estado expuesto al asbesto u otras toxinas y si ha sido fumador. En muchos pacientes, los síntomas están presentes durante bastante tiempo antes del diagnóstico. Entre las características clínicas más habituales de la FPI se incluyen:
En el examen físico se puede encontrar:
Ruidos respiratorios anormales llamados crepitaciones.
.En muchos pacientes, los síntomas están presentes durante bastante tiempo antes del diagnóstico. Entre las características clínicas más habituales de la FPI se incluyen:
- Edad superior a los 50 años
- Dolor torácico (ocasionalmente)
- Tos (por lo general seca) improductiva en esfuerzo
- Disminución de la tolerancia a la actividad
- Disnea de esfuerzo progresiva (dificultad al respirar durante una actividad ó ejercicio) este síntoma dura meses o años y con el tiempo también puede ocurrir en reposo
- Estertores inspiratorios secos y bibasilares en la auscultación (Con sonido crepitante en los pulmones durante la inspiración).
- Coloración azulada en la piel (cianosis) alrededor de la boca o en las uñas, debido a la insuficiencia de oxígeno (con la enfermedad avanzada).
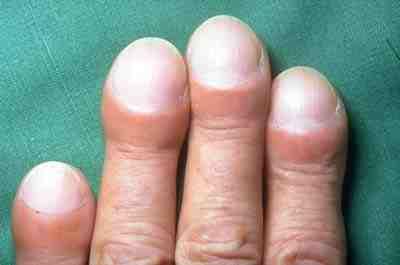
- Dedos hipocráticos (véase la imagen)
- Resultados anómalos en las pruebas pulmonares funcionales, con indicios de obstrucción y alteración en el intercambio gaseoso.
Estas características se deben a la deficiencia crónica de oxígeno en sangre y pueden estar presentes en una amplia variedad de otros trastornos pulmonares, no siendo específicas de la FPI. Sin embargo, debe considerarse la presencia de FPI en todos aquellos pacientes con disnea crónica de esfuerzo inexplicable que cursen con tos, estertores inspiratorios bibasilares o dedos hipocráticos.
La valoración de los estertores “crepitantes” en la auscultación pulmonar es una forma práctica de mejorar el diagnóstico precoz de la FPI. El médico puede reconocer fácilmente los sutiles estertores característicos de la FPI.
Si se hallan sutiles estertores bilaterales durante todo el periodo inspiratorio que persisten tras varias inspiraciones profundas, y si continúan presentes en diferentes momentos con varias semanas de diferencia en un sujeto con edad ≥60 años, debería sospecharse la presencia de FPI y considerarse la opción de realizar una TAC del tórax, la cual es más sensible que una radiografía del tórax. Debido a que los estertores no son un signo específico de la FPI, se debe realizar un proceso diagnóstico más exhaustivo.
El reconocimiento de estos síntomas requiere una serie de investigaciones adicionales con objeto de diagnosticar la FPI.
 EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG |
Los exámenes que ayudan a diagnosticar la fibrosis pulmonar idiopática son, entre otros, los siguientes:
- Hemograma
- Química
- Anticuerpos para enfermedades del tejido conectivo como artritis reumatoidea, lupus o esclerodermia
- Antígenos cancergenos
- Biopsia de pulmón abierto (quirúrgica)
Los exámenes que ayudan a diagnosticar la fibrosis pulmonar idiopática son, entre otros, los siguientes:
- Broncoscopia
- Pruebas de la función pulmonar
- Prueba de la caminata de 6 minutos
- Biopsia de pulmón abierto (quirúrgica)
Las radiografías de tórax resultan útiles para el seguimiento sistemático de los pacientes con FPI. Desafortunadamente, las radiografías de tórax estándar no ofrecen un diagnóstico, pero pueden revelar una reducción del volumen pulmonar, normalmente con marcas intersticiales reticulares prominentes cerca de las bases pulmonares.1
La evaluación radiológica mediante TCAR es un elemento esencial en la ruta diagnóstica de la FPI. La TCAR se lleva a cabo con una gammacámara tomográfica axial computarizada convencional sin la inyección de agentes de contraste. Los cortes de evaluación son muy finos (1–2 mm).
La TCAR típica del tórax en un paciente con FPI muestra los cambios fibróticos en ambos pulmones, con una predilección por las bases pulmonares y la periferia.De acuerdo con las directrices conjuntas ATS/ERS/JRS/ALAT de 2011, la TCAR es un componente esencial en la ruta de diagnóstico de la FPI, la cual puede identificar NIU por la presencia de:
- Opacidades reticulares, a menudo asociadas con bronquiectasia por tracción
- Panalización manifestada como espacios quísticos agrupados, normalmente con diámetros de entre 3 y 10 mm, pero a veces de mayor tamaño. Normalmente subpleurales y que se caracterizan por paredes bien definidas y dispuestas en, al menos dos líneas. Generalmente, una línea de quiste no es suficiente para definir la panalización.
- Las opacidades con aspecto de vidrio deslustrado son comunes pero menos amplias que la reticulación
- Distribución predominantemente basal y periférica aunque, a menudo, con patrón parcheado.
 |
Imágenes tomográficas computarizadas de alta resolución del tórax de un paciente con FPI. Las características principales son de una amplia reticulación con panalización, con patrón periférico y predominantemente basal  EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG |
La mayor parte de estos pacientes necesitan mantenimiento de sostén debido a la evolución crónica de su cuadro y el mal pronóstico de los mismos. Se recomienda dar oxigenación por cánula y si se tiene broncodilatador con esteroide suministrar nebulizacion en el recorrido hasta llegar al centro de tercer nivel. Se debe estar preparado para dar la Reanimación cardiopulmonar en caso de ser necesario.
► Enfoques terapéuticos
En los ensayos clínicos de nuevos fármacos (etanercept, IFNx, bosentan, mesilato de imatinib) en pacientes con FPI con deterioro funcional leve a moderado no se han observado beneficios significativos. En general, los pacientes con FPI con deterioro funcional moderado a grave y comorbilidades asociadas (hipertensión pulmonar) han sido excluidos de los ensayos. En consecuencia, muchos pacientes atendidos en la práctica clínica no han sido estudiados. Están en desarrollo vario ensayos clínicos.
Con varias terapias nuevas, hay evidencia que sugiere beneficiosos clínicos en pacientes con FPI. La N-acetilcisteína, un antioxidante que se utiliza combinado con prednisona y azatioprina, reduce la tasa de disminución de la capacidad vital forzada y la capacidad de difusión del monóxido de carbono después de 12 meses de tratamiento. Sin embargo, los cambios observados son de significado clínico incierto. La pirfenidona, una compuesto nuevo que inhibe el TGF in vitro, disminuyó la tasa de declinación de la capacidad vital y aumentó el tiempo de supervivencia libre de progresión. Los datos combinados de 2 ensayos de FPI simultáneos en fase 3 (CAPACITY1 y 2) indican que la variación media de la capacidad vital forzada y la distancia en la caminata de 6 minutos disminuyeron en el grupo tratado con pirfenidona.
En un ensayo no ciego de FPI, los pacientes tratados con prednisolona y anticoagulantes (coumadin en pacientes ambulatorios y dalteparina intravenosa al ingresar al hospital) mejoraron la supervivencia global a los 3 años y la mortalidad asociada con la exacerbación aguda (63% vs. 35%) disminuyó en comparación con los pacientes tratados con prednisolona sola. Un estudio con warfarina para pacientes con FPI fue suspendido por la elevada posibilidad de que su efecto no fuera muy superior al del placebo.
► Trasplante de pulmón
El trasplante de pulmón es el único tratamiento que prolonga la supervivencia en la FPI avanzada, aunque la supervivencia postrasplante a los 5 años es de alrededor del 44%. El momento apropiado para remitir al paciente para el trasplante es muy controvertido dado el curso variable de la FPI y la falta de medidas de pronóstico validadas. Los pacientes con FPI suelen consultar tarde en el curso de su enfermedad y muchos mueren antes de recibir un trasplante. Por lo tanto, sería razonable derivar a los pacientes para trasplante apenas realizado el diagnóstico.
► Tratamiento con células madre
Tanto las células embrionarias como las células madre derivadas del tejido adulto pueden participar en la regeneración y reparación de los órganos de los adultos enfermos, incluyendo los pulmones. El principal problema después de la lesión es restablecer la integridad y la organización funcional de la capa epitelial y las unidades alveolocapilares. Este proceso probablemente se produce durante la reparación normal por la migración y la difusión de las células progenitoras circulantes vecinas y recién reclutadas que proliferan y se diferencian fenotípicamente para cubrir las superficies desnudas.
Las células madre mesenquimales son una perspectiva prometedora para la regeneración tisular. En los ratones con lesión pulmonar por bleomicina, estas células emigran al pulmón, adoptan un fenotipo similar al epitelio y reducen la fibrosis. Por otra parte, después de la inyección intratraqueal de bleomicina, las células progenitoras epiteliales positivas para prominina 1 CD133 coexpresan marcadores de células madre y hematopoyéticas y se injertan en los pulmones, se diferencian en neumocitos tipo II y protegen contra la fibrosis inducida por bleomicina. Más recientemente, en el mismo modelo de ratón se estudió el potencial terapéutico de las CEA tipo II derivadas de células madre embrionarias humanas. Estas células diferenciadas se diferenciaron en neumocitos tipo I y anularon la respuesta inflamatoria y fibrótica. Por desgracia, casi todos los experimentos se han hecho en el modelo de pulmón lesionado por bleomicina, en el que la inflamación y la fibrosis son escasas y espontáneamente reversibles y no representan a la naturaleza progresiva y letal de la FPI.
Otra área de crecimiento de la investigación es el pulmón de bioingeniería usando tejido pulmonar en el que se cambiaron las células naturales por células del epitelio pulmonar neonatal y del endotelio microvascular del pulmón. Cuando se trasplantaron, estos pulmones fueron efectivos para el intercambio de oxígeno y del dióxido de carbono. Aunque se está lejos de la bioingeniería humana del pulmón, este enfoque es alentador.
Los pacientes con exacerbaciones agudas suelen tratarse con antibióticos de amplio espectro y corticoides. A menudo es necesaria la ventilación mecánica, pero por lo general no tiene buenos resultados, con una tasa elevada de mortalidad hospitalaria. Entre los pacientes que sobreviven y son dados de alta del hospital, la recurrencia es común y generalmente fatal. Los pacientes con FPI e hipertensión pulmonar tienen mayor mortalidad. En consecuencia, la terapia contra la hipertensión arterial pulmonar brinda beneficios. El sildenafil, un bloqueante de la fosfodiesterasa 5 en las áreas de pulmón bien ventiladas, reduce la resistencia vascular pulmonar y mejora el intercambio gaseoso en los pacientes con fibrosis pulmonar grave. El sildenafil puede mejorar mucho la disnea y la calidad de vida de los pacientes con FPI.
Aunque la asociación patogénica potencial entre la microaspiración crónica y la FPI sigue sin estar clara, hay algunas pruebas que apoyan el manejo del reflujo gastroesofágico. Sin embargo, se necesitan más estudios para comprobar si el tratamiento agresivo de esta enfermedad crónica es capaz de mejorar o detener su progresión. Teniendo en cuenta la relación entre el envejecimiento y la FPI, los médicos deben prestar mayor atención a las comorbilidades geriátricas y al manejo de los síntomas para complementar las nuevas terapias modificadoras de la enfermedad y mejorar la calidad de vida.
La rehabilitación pulmonar, los programas educativos y la formación de grupos de apoyo pueden ayudar a los pacientes a respirar mejor y realizar a sus actividades de la vida diaria con menos dificultad respiratoria. Para tratar la hipoxemia que tiende a empeorar con el ejercicio suele ser necesaria la oxigenoterapia.
La evolución clínica de la FPI puede ser impredecible.1 31 32 El avance de la FPI se asocia con un periodo de supervivencia medio estimado de entre 2 y 5 años tras su diagnóstico.1 La supervivencia a 5 años para la FPI se estima entre el 20 y el 40 %,32 un índice de mortalidad superior al de varias neoplasias malignas, incluido el cáncer colorrectal, mieloma múltiple y el cáncer de vejiga.31 32
Recientemente se ha propuesto un sistema de indexación y clasificación multidimensional para predecir la mortalidad por FPI.33 El nombre del índice es GAP y se basa en las iniciales en inglés de gender (sexo) [G], age (edad) [A], y dos variables pulmonares fisiológicas [P] (FVC y DLCO] que habitualmente se miden en la práctica médica para predecir la mortalidad de la FPI. El grado más alto del GAP (Grado III) se ha asociado con un 39 % de riesgo de mortalidad en 1 año.33 Este modelo también se ha evaluado en la FPI y otras NI, habiéndose mostrado unos buenos resultados en la predicción de la mortalidad en todos los subtipos principales de NI. Se ha desarrollado un índice NI-GAP modificado para su aplicación a los diferentes subtipos de NI con objeto de ofrecer estimaciones de supervivencia específicas de la enfermedad.34 En los pacientes de NI, la tasa de mortalidad general a 5 años es elevada, pero la tasa anual de mortalidad por cualquier causa en pacientes con alteración pulmonar leve o moderada, es relativamente baja. Este es el motivo por el que normalmente, en los estudios clínicos anuales de tratamientos para la FPI, se mide el cambio en la función pulmonar (FVC), en lugar de la supervivencia.35
Además de los parámetros clínicos y fisiológicos para predecir la posible rapidez en evolución de los pacientes con FPI, las características genéticas y moleculares se asocian también con la mortalidad por FPI. Por ejemplo, se ha demostrado que los pacientes de FPI con un genotipo específico en el polimorfismo del gen MUC5B de la mucina (véase información anterior) experimentan un declive más lento de la FVC, así como una tasa de supervivencia notablemente mayor.36 37 Aunque dichos datos resultan interesantes desde el punto de vista científico, todavía resulta imposible aplicar un modelo pronóstico basado en genotipos específicos en la práctica médica rutinaria.
Comparación de la tasa de supervivencia a 5 años para la FPI y varias neoplasias malignas comunes. Adaptado de Bjoraker y cols 1998.  EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG |
► Conclusiones y enfoques a futuro
En la última década, el conocimiento de la patogenia de la fibrosis pulmonar idiopática cambió considerablemente. Antes se pensaba que se generaba por un proceso inflamatorio crónico, ahora se sabe que es causada por el trastorno de las células epiteliales alveolares, la expansión de las poblaciones de fibroblastos y miofibroblastos pulmonares y la secreción de cantidades excesivas de componentes de la matriz extracelular, que produce cicatrización y destrucción de la arquitectura pulmonar.
Factores genéticos aumentan de manera importante el desarrollo de la fibrosis pulmonar idiopática en personas con la enfermedad familiar o esporádica. Sin embargo, la mayoría de los casos de fibrosis pulmonar idiopática no tienen un vínculo genético evidente.
Posibles explicaciones son las limitaciones de las pruebas genotípicas disponibles y la insuficiente consideración de los factores ambientales. Puede haber cantidades mucho mayores de variantes comunes de menor efecto o identificadas; variantes raras con efectos mayores o variantes estructurales (inserción, deleción, duplicación, traslocación o inversión de segmentos de DNA) y dificultades en la detección de interacciones gen-gen.
El empleo de tecnologías de secuenciación de nueva generación probablemente aumentará la cantidad de variantes genéticas asociadas con la fibrosis pulmonar idiopática. Aunque se apliquen estándares de calidad rigurosos a los datos del genotipo, la delineación exacta y precisa del fenotipo blanco es problemática en la fibrosis pulmonar idiopática porque se han propuesto varios fenotipos diferentes y es probable que existan muchos más tipos. El fenotipo blanco está aún menos definido en las enfermedades del tejido conjuntivo o la fibrosis pulmonar relacionada con la sarcoidosis.
Otra explicación de la ausencia de determinantes genéticos para explicar la posibilidad de heredar la fibrosis pulmonar idiopática es la participación de mecanismos epigenéticos (eg, cambios heredables en la expresión de genes causados por factores distintos a los cambios en la secuencia de ADN).
Los cambios epigenéticos pueden activar o desactivar los genes para determinar qué proteínas se transcriben y para permitir el desarrollo y la diferenciación, la homeostasis de los tejidos sanos y la capacidad del huésped de responder al estrés. Es probable que participen mecanismos epigenéticos en la expresión de los genes en la fibrosis pulmonar idiopática, en especial dada la asociación de ésta con el tabaquismo.
La epigenética podría ser el vínculo que falta entre la exposición ambiental en individuos genéticamente predispuestos y el desarrollo de fibrosis pulmonar idiopática. La expresión de conglomerados de miR-17-92, que es importante para mantener la homeostasis de las células epiteliales pulmonares y en la reparación pulmonar, está disminuida en muestras de biopsia pulmonar y en fibroblastos pulmonares de pacientes con fibrosis pulmonar idiopática. El silenciamiento epigenético del conglomerado miR-17-92 se produce debido al aumento de la metilación del ADN, de modo que dirigir el tratamiento a ésta es una estrategia terapéutica posible.
Aun se progresó mucho en el conocimiento de la patobiología de la fibrosis pulmonar idiopática en la última década, aún no se conocen por completo las causas de la forma más letal de fibrosis pulmonar, la fibrosis pulmonar idiopática. Tampoco hay enfoques terapéuticos universalmente eficaces, aunque se hicieron algunos progresos con tratamientos que reducen la rapidez de la progresión de la enfermedad.
No obstante, los estudios genéticos ofrecen la esperanza de identificar con más precisión a los individuos en riesgo de fibrosis pulmonar idiopática; de definir la patogenia de la enfermedad y tarde o temprano de crear tratamientos dirigidos selectivamente a las vías genéticas y bioquímicas clave de la enfermedad.
►Artículo ➔ Noticia ➲ Tema básico ➜ Editorial
1 Eckes B, Kessler D, Aumailley M, Krieg T. Interactions of fibroblasts with the extracellular matrix: implications for the understanding of fi brosis. Springer Semin Immunopathol 2000; 21: 415–29.
2 Wynn TA. Cellular and molecular mechanisms of fi brosis. J Pathol 2008; 214: 199–210.
3 Macneal K, Schwartz DA. The genetic and environmental causes of pulmonary fi brosis. Proc Am Thorac Soc 2012; 9: 120–25.
4 Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classifi cation of the idiopathic interstitial pneumonias.
5 Bjoraker JA, Ryu JH, Edwin MK, et al. Prognostic significance of histopathologic subsets in idiopathic pulmonary fi brosis. Am J Respir Crit Care Med 1998; 157: 199–203.
6 Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fi brosis. Am J Respir Crit Care Med 2011; 183: 431–40.
7 Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fi brosis (CAPACITY): two randomized trials. Lancet 2011; 377: 1760–69.
8 Richeldi L, Costabel U, Selman M, et al. Effi cacy of a tyrosine kinase inhibitor in idiopathic pulmonary fi brosis. N Engl J Med 2011; 365: 1079–87.
9 Spagnolo P, Luppi F, Cerri S, Richeldi L. Genetic testing in diffuse parenchymal lung disease. Orphanet J Rare Dis 2012; 7: 79.
10 Armanios MY, Chen JJ, Cogan JD, et al. Telomerase mutations in families with idiopathic pulmonary fi brosis. N Engl J Med 2007; 356: 1317–26.
11 Depinho RA, Kaplan KL. The Hermansky-Pudlak syndrome: report of three cases and review of pathophysiology and management considerations. Medicine 1985; 64: 192–202.
12 Javaheri S, Lederer DH, Pella JA, Mark GJ, Levine BW. Idiopathic pulmonary fibrosis in monozygotic twins: the importance of genetic predisposition. Chest 1980; 78: 591–94.
13 Hughes EW. Familial interstitial pulmonary fi brosis. Thorax 1964; 19: 515–25.
14 Bitterman PB, Rennard SI, Keogh BA, Wewers MD, Adelberg S, Crystal RG. Familial idiopathic pulmonary fibrosis: evidence of lung inflammation in unaffected members. N Engl J Med 1986; 314: 1343–47.
15 Steele MP, Speer MC, Loyd JE, et al. Clinical and pathological features of familial interstitial pneumonia. Am J Respir Crit Care Med 2005; 172: 1146–52.
16 Hodgson U, Laitinen T, Tukiainen P. Nationwide prevalence of sporadic and familial idiopathic pulmonary fi brosis: evidence of founder effect among multiplex families in Finland. Thorax 2002; 57: 338–42.
17 Peabody JW, Peabody JW Jr, Hayes EW, et al. Idiopathic pulmonary fibrosis; its occurrence in identical twin sisters. Dis Chest 1950; 18: 330–44.
18 MacMillan JM. Familial pulmonary fi brosis. Dis Chest 1951; 20: 426–36.
19 van Moorsel CH, van Oosterhout MF, Barlo NP, et al. SFTPC mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a Dutch cohort. Am J Respir Crit Care Med 2010; 182: 1419–25.
20 Lawson WE, Loyd JE, Degryse AL. Genetics in pulmonary fi brosis—familial cases provide clues to the pathogenesis of idiopathic pulmonary fibrosis. Am J Med Sci 2011; 341: 439–43.
21 Lee H, Ryu JH, Wittmer MH, et al. Familial idiopathic pulmonary fibrosis: clinical features and outcome. Chest 2005; 127: 2034–41.
22 Marshall RP, Puddicombe A, Cookson WO, et al. Adult familial cryptogenic fi brosing alveolitis in the United Kingdom. Thorax 2000; 55: 143–46.
23 Lee HY, Seo JB, Steele MP, et al. High-resolution CT scan findings in familial interstitial pneumonia do not conform to those of idiopathic interstitial pneumonia. Chest 2012; 142: 1577–83.
24 Nogee LM, Dunbar AE 3rd, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med 2001; 344: 573–79.
25 Thomas AQ, Lane K, Phillips J 3rd, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecifi c interstitial pneumonitis in one kindred. Am J Respir Crit Care Med 2002; 165: 1322–28.
26 Whitsett JA, Weaver TE. Hydrophobic surfactant proteins in lung function and disease. N Engl J Med 2002; 347: 2141–48.
27 Mulugeta S, Nguyen V, Russo SJ, Muniswamy M, Beers MF. A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation. Am J Respir Cell Mol Biol 2005; 32: 521–30.
28 Lawson WE, Crossno PF, Polosukhin VV, et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol 2008;
294: L1119–26.
29 Korfei M, Ruppert C, Mahavadi P, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2008; 178: 838–46.
30 Wang Y, Kuan PJ, Xing C, et al. Genetic defects in surfactant protein A2 are associated with pulmonary fi brosis and lung cancer. Am J Hum Genet 2009; 84: 52–59.
31 Maitra M, Wang Y, Gerard RD, Mendelson CR, Garcia CK. Surfactant protein A2 mutations associated with pulmonary fi brosis lead to protein instability and endoplasmic reticulum stress. J Biol Chem 2010; 285: 22103–13.
32 Calado RT, Young NS. Telomere diseases. N Engl J Med 2009; 361: 2353–65.
33 Nelson ND, Bertuch AA. Dyskeratosis congenita as a disorder of telomere maintenance. Mutat Res 2012; 730: 43–51.
34 Dokal I. Dyskeratosis congenita in all its forms. Br J Haematol 2000; 110: 768–79.
35 Vulliamy T, Marrone A, Szydlo R, et al. Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nat Genet 2004; 36: 447–49.
36 Vulliamy T, Marrone A, Goldman F, et al. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature 2001; 413: 432–35.
37 Armanios M, Chen JL, Chang YP, et al. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc Natl Acad Sci USA 2005; 102: 15960–64.
38 Vulliamy T, Dokal I. Dyskeratosis congenita. Semin Hematol 2006; 43: 157–66.
39 Tsakiri KD, Cronkhite JT, Kuan PJ, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci USA 2007; 104: 7552–57.
40 Cronkhite JT, Xing C, Raghu G, et al. Telomere shortening in familial and sporadic pulmonary fi brosis. Am J Respir Crit Care Med 2008; 178: 729–37.
41 Alder JK, Chen JJ, Lancaster L, et al. Short telomeres are a risk factor for idiopathic pulmonary fi brosis. Proc Natl Acad Sci USA 2008; 105: 13051–56.
42 Armanios M. Telomerase and idiopathic pulmonary fibrosis. Mutat Res 2012; 730: 52–58.
43 Tsuji T, Aoshiba K, Nagai A. Alveolar cell senescence in patients with pulmonary emphysema. Am J Respir Crit Care Med 2006; 174: 886–93.
44 Valdes AM, Andrew T, Gardner JP, et al. Obesity, cigarette smoking, and telomere length in women. Lancet 2005; 366: 662–64.
45 O’Sullivan JN, Bronner MP, Brentnall TA, et al. Chromosomal instability in ulcerative colitis is related to telomere shortening. Nat Genet 2002; 32: 280–84.
46 Risques RA, Lai LA, Brentnall TA, et al. Ulcerative colitis is a disease of accelerated colon aging: evidence from telomere attrition and DNA damage. Gastroenterology 2008; 135: 410–18.
47 Armanios M. Syndromes of telomere shortening. Annu Rev Genomics Hum Genet 2009; 10: 45–61.
48 Gribbin J, Hubbard R, Smith C. Role of diabetes mellitus and gastro-oesophageal reflux in the aetiology of idiopathic pulmonary fibrosis. Respir Med 2009; 103: 927–31.
49 International HapMap Consortium. A haplomap type of the human genome. Nature 2005; 437: 1299–320.
50 Wang WY, Barratt BJ, Clayton DG, Todd JA. Genome-wide association studies: theoretical and practical concerns. Nat Rev Genet 2005; 6: 109–18.
51 Manolio TA. Genomewide association studies and assessment of the risk of disease. N Engl J Med 2010; 363: 166–76.
52 Hodgson U, Pulkkinen V, Dixon M, et al. ELMOD2 is a candidate gene for familial idiopathic pulmonary fibrosis. Am J Hum Genet 2006; 79: 149–54.
53 Pulkkinen V, Bruce S, Rintahaka J, et al. ELMOD2, a candidate gene for idiopathic pulmonary fi brosis, regulates antiviral responses. FASEB J 2010; 24: 1167–77.
54 Mushiroda T, Wattanapokayakit S, Takahashi A, et al. A genomewide association study identifi es an association of a common variant in TERT with susceptibility to idiopathic pulmonary fi brosis. J Med Genet 2008; 45: 654–56. 12
55 Korthagen NM, van Moorsel CH, Barlo NP, Kazemier KM, Ruven HJ, Grutters JC. Association between variations in cell cycle genes and idiopathic pulmonary fi brosis. PLoS One 2012; 7: e30442
56 Xue J, Gochuico BR, Alawad AS, et al. The HLA class II allele DRB1*1501 is over-represented in patients with idiopathic pulmonary fi brosis. PLoS One 2011; 6: e14715.
57 Whyte M, Hubbard R, Meliconi R, et al. Increased risk of fibrosing alveolitis associated with interleukin-1 receptor antagonist and tumor necrosis factor-alpha gene polymorphisms. Am J Respir Crit Care Med 2000; 162: 755–58.
58 Barlo NP, van Moorsel CH, Korthagen NM, et al. Genetic variability in the IL1RN gene and the balance between interleukin (IL)-1 receptor agonist and IL-1β in idiopathic pulmonary fi brosis. Clin Exp Immunol 2011; 166: 346–51.
59 Korthagen NM, van Moorsel CH, Kazemier KM, Ruven HJ, Grutters JC. IL1RN genetic variations and risk of IPF: a meta-analysis and mRNA expression study. Immunogenetics 2012; 64: 371–77.
60 Ahn MH, Park BL, Lee SH, et al. A promoter SNP rs4073T>A in the common allele of the interleukin 8 gene is associated with the development of idiopathic pulmonary fi brosis via the IL-8 protein enhancing mode. Respir Res 2011; 12: 73.
61 Seibold MA, Wise AL, Speer MC, et al. A common MUC5B promoter polymorphism and pulmonary fi brosis. N Engl J Med 2011; 364: 1503–12.
62 Zhang Y, Noth I, Garcia GN, Kaminski N. A variant in the promoter of MUC5B and idiopathic pulmonary fi brosis. N Engl J Med 2011; 364: 1576–77.
63 Stock CJ, Sato H, Fonseca C, et al. Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fi brosis but not with development of lung fi brosis in systemic sclerosis or sarcoidosis. Thorax 2013; 68: 436–41.
64 Peljto AL, Zhang Y, Fingerlin TE, et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA 2013; 309: 2232–39.
65 Xaubet A, Marin-Arguedas A, Lario S, et al. Transforming growth factor-b1 gene polymorphisms are associated with disease progression in idiopathic pulmonary fi brosis. Am J Respir Crit Care Med 2003; 168: 431–35.
66 Son JY, Kim SY, Cho SH, et al. TGF-β1 T869C polymorphism may affect susceptibility to idiopathic pulmonary fi brosis and disease severity. Lung 2013; 191: 199–205.
67 Noth I, Zhang Y, Ma SF, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med 2013; 1: 309–17.
68 Fingerlin TE, Murphy E, Zhang W, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fi brosis. Nat Genet 2013; 45: 613–20.
69 Nekrasova O, Green KJ. Desmosome assembly and dynamics. Trends Cell Biol 2013; 23: 537–46.
70 Borie R, Crestani B, Dieude P, et al. The MUC5B variant is associated with idiopathic pulmonary fibrosis but not with systemic sclerosis interstitial lung disease in the European Caucasian population. PLoS One 2013; 8: e70621.
71 Peljto AL, Steele MP, Fingerlin TE, et al. The pulmonary fibrosis associated
MUC5B promoter polymorphism does not influence the development of interstitial pneumonia in systemic sclerosis. Chest 2012; 142: 1584–88.
72 Odoardi F, Sie C, Streyl K, et al. T cells become licensed in the lung to enter the central nervous system. Nature 2012; 488: 675–79.
73 Fischer A, du Bois RM. Interstitial lung disease in connective tissue disorders. Lancet 2012; 380: 689–98.
74 Park JH, Kim DS, Park IN, et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med 2007; 175: 705–11.
75 Nalysnyk L, Cid-Ruzafa J, Rotella P, Essere D. Incidence and prevalence of idiopathic pulmonary fi brosis: review of the literature. Eur Respir Rev 2012; 21: 355–61.
76 Klareskog L, Stolt P, Lundberg K, et al. A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modifi ed by citrullination. Arthritis Rheum 2006; 54: 38–46.
77 Makrygiannakis D, Hermansson M, Ulfgren AK, et al. Smoking increases peptidylarginine deiminase 2 enzyme expression in human lungs and increases citrullination in BAL cells. Ann Rheum Dis 2008; 67: 1488–92.
78 Reynisdottir G, Karimi R, Joshua V, et al. Structural lung changes and local anti-citrulline immunity are early features of anti citrullinated proteins antibodies positive rheumatoid arthritis. Arthritis Rheum 2013; published online Oct 21. DOI:10.1002/art.38201.
79 Rantapaa-Dahlqvist S, de Jong BA, Berglin E, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum 2003; 48: 2741–49.
80 Klareskog L, Catrina AI, Paget S. Rheumatoid arthritis. Lancet 2009; 373: 659–72.
81 Kim EJ, Collard HR, King TE Jr. Rheumatoid arthritis-associated interstitial lung disease: the relevance of histopathologic and radiographic pattern. Chest 2009; 136: 1397–405.
82 Mori S, Cho I, Koga Y, Sugimoto M. Comparison of pulmonary abnormalities on high-resolution computed tomography in patients with early versus longstanding rheumatoid arthritis. J Rheumatol 2008; 35: 1513–21.
83 Turesson C. Extra-articular rheumatoid arthritis. Curr Opin Rheumatol 2013; 25: 360–66.
84 Charles PJ, Sweatman MC, Markwick JR, Maini RN. HLA-B40: a marker for susceptibility to lung disease in rheumatoid arthritis. Dis Markers 1991; 9: 97–101.
85 Sugiyama Y, Ohno S, Kano S, Maeda H, Kitamura S. Diffuse panbronchiolitis and rheumatoid arthritis: a possible correlation with HLA-B54. Intern Med 1994; 33: 612–14.
86 Scott TE, Wise RA, Hochberg MC, Wigley FM. HLA-DR4 and pulmonary dysfunction in rheumatoid arthritis. Am J Med 1987; 82: 765–71.
87 Migita K, Nakamura T, Koga T, Eguchi K. HLA-DRB1 alleles and rheumatoid arthritis-related pulmonary fibrosis. J Rheumatol 2010; 37: 205–07.
88 Furukawa H, Oka S, Shimada K, et al. Association of human leukocyte antigen with interstitial lung disease in rheumatoid arthritis: a protective role for shared epitope. PLoS One 2012; 7: e33133.
89 Fischer A, Solomon JJ, du Bois RM, et al. Lung disease with anti-CCP antibodies but not rheumatoid arthritis or connective tissue disease. Respir Med 2012; 106; 1040–47.
90 Sharif R, Mayes MD, Tan FK, et al. IRF5 polymorphism predicts prognosis in patients with systemic sclerosis. Ann Rheum Dis 2012; 71: 1197–202.
91 Assassi S, Del Junco D, Sutter K, et al. Clinical and genetic factors predictive of mortality in early systemic sclerosis. Arthritis Rheum 2009; 61: 1403–11.
92 Goh NS, Desai SR, Veeraraghavan S, et al. Interstitial lung disease in systemic sclerosis: a simple staging system. Am J Respir Crit Care Med 2008; 177: 1248–54.
93 Steen VD, Powell DL, Medsger TA Jr. Clinical correlations and prognosis based on serum autoantibodies in patients with progressive systemic sclerosis. Arthritis Rheum 1988; 31: 196–203.
94 Vlachoyiannopoulos PG, Dafni UG, Pakas I, Spyropoulou-Vlachou M, Stavropoulos-Giokas C, Moutsopoulos HM. Systemic scleroderma in Greece: low mortality and strong linkage with HLA-DRB1*1104 allele. Ann Rheum Dis 2000; 59: 359–67.
95 Gilchrist FC, Bunn C, Foley PJ, et al. Class II HLA associations with autoantibodies in scleroderma: a highly significant role for HLA-DP. Genes Immun 2001; 2: 76–81.
96 Tikly M, Rands A, McHugh N, Wordsworth P, Welsh K. Human leukocyte antigen class II associations with systemic sclerosis in South Africans. Tissue Antigens 2004; 63: 487–90.
97 Radstake TR, Gorlova O, Rueda B, et al. Genome-wide association study of systemic sclerosis identifi es CD247 as a new susceptibility locus. Nat Genet 2010; 42: 426–29.
98 Black CM, Silman AJ, Herrick AI, et al. Interferon-alpha does not improve outcome at one year in patients with diff use cutaneous scleroderma: results of a randomized, double-blind, placebo-controlled trial. Arthritis Rheum 1999; 42: 299–305.
99 Koumakis E, Bouaziz M, Dieude P, et al. A regulatory variant in CCR6 is associated with anti-topoisomerase positive systemic sclerosis susceptibility. Arthritis Rheum 2013; 65: 3202–08.
100 Dieude P, Guedj M, Wipff J, et al. NLRP1 influences the systemic sclerosis phenotype: a new clue for the contribution of innate immunity in systemic sclerosis-related fibrosing alveolitis pathogenesis. Ann Rheum Dis 2011; 70: 668–74.
101 Abehsera M, Valeyre D, Grenier P, Jaillet H, Battesti JP, Brauner MW. Sarcoidosis with pulmonary fibrosis: CT patterns and correlation with pulmonary function. AJR Am J Roentgenol 2000; 174: 1751–57.
102 Rybicki BA, Iannuzzi MC, Frederick MM, et al. Familial aggregation of sarcoidosis. A case-control etiologic study of sarcoidosis (ACCESS). Am J Respir Crit Care Med 2001; 164: 2085–91.
103 Sverrild A, Backer V, Kyvik KO, et al, Heredity in sarcoidosis: a registry-based twin study. Thorax 2008; 63: 894–96.
104 Judson M, Hirst K, Ivengar SK, et al. Comparison of sarcoidosis phenotypes among aff ected African-American siblings. Chest 2006; 130: 855–62.
105 Spagnolo P, Grunewald J. Recent advances in the genetics of sarcoidosis. J Med Genet 2013; 50: 290–97.
106 Grunewald J, Eklund A. Lofgren’s syndrome: human leukocyte antigen strongly infl uences the disease course. Am J Respir Crit Care Med 2009; 179: 307–12.
107 Li Y, Wollnik B, Pabst S, et al. BTNL2 gene variant and sarcoidosis. Thorax 2006; 61: 273–74.
108 Wijnen PA, Voorter CE, Nelemans PJ, Verschakelen JA, Bekers O, Drent M. Butyrophilin-like 2 in pulmonary sarcoidosis: a factor for susceptibility and progression? Hum Immunol 2011; 72: 342–47.
109 Spagnolo P, Sato H, Grutters JC, et al. Analysis of BTNL2 genetic polymorphisms in British and Dutch patients with sarcoidosis. Tissue Antigens 2007; 70: 219–27.
110 Heron M, van Moorsel CH, Grutters JC, et al. Genetic variation in GREM1 is a risk factor for fi brosis in pulmonary sarcoidosis. Tissue Antigens 2010; 77: 112–17.
111 Sato H, Williams HR, Spagnolo P, et al. CARD15/NOD2 polymorphisms are associated with severe pulmonary sarcoidosis. Eur Respir J 2010; 35: 324–30.
112 Kruit A, Grutters JC, Ruven HJ, et al. Transforming growth factor-beta gene polymorphisms in sarcoidosis patients with and without fi brosis. Chest 2006; 129: 1584–91.
113 Lawson WE, Polosukhin VV, Stathopoulos GT, et al. Increased and prolonged pulmonary fi brosis in surfactant protein C–defi cient mice following intratracheal bleomycin. Am J Pathol 2005; 167: 1267–77.
114 Noble PW, Barkauskas CE, Jiang D. Pulmonary fibrosis: patterns and perpetrators. J Clin Invest 2012; 122: 2756–62.
115 Parker D, Prince A. Innate immunity in the respiratory epithelium. Am J Respir Cell Mol Biol 2011; 45: 189–201.
116 Plantier L, Crestani B, Wert SE, et al. Ectopic respiratory epithelial cell differentiation in bronchiolised distal airspaces in idiopathic pulmonary fi brosis. Thorax 2011; 66: 651–57.
117 Seibold MA, Smith RW, Urbanek C, et al. The idiopathic pulmonary fi brosis honeycomb cyst contains a mucocilary pseudostratified epithelium. PLoS One 2013; 8: e58658.
118 Boucher RC. Idiopathic pulmonary fibrosis—a sticky business. N Engl J Med 2011; 364: 1560–61.
119 Martino MB, Jones L, Brighton B, et al. The ER stress transducer IRE1β is required for airway epithelial mucin production. Mucosal Immunol 2013; 6: 639–54.
120 Vuga LJ, Ben-Yehudah A, Kovkarova-Naumovski E, et al. WNT5A is a regulator of fi broblast proliferation and resistance to apoptosis. Am J Respir Cell Mol Biol 2009; 41: 583–89.
121 Gunther A, Korfei M, Mahavadi P, von der Beck D, Ruppert C, Markart P. Unravelling the progressive pathophysiology of idiopathic pulmonary fibrosis. Eur Respir Rev 2012; 21: 152–60.
122 Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med 2011; 208: 1339–50.
123 Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol 2003; 200: 500–03.
124 Wells AU, Cullinan P, Hansell DM, et al. Fibrosing alveolitis associated with systemic sclerosis has a better prognosis than lone cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med 1994; 149: 1583–90.
125 Koli K, Myllarniemi M, Vuorinen K, et al. Bone morphogenetic protein-4 inhibitor gremlin is overexpressed in idiopathic pulmonary fibrosis. Am J Pathol 2006; 169: 61–71.
126 Raghu G, Anstrom KJ, King TE Jr, et al, for the Idiopathic Pulmonary Fibrosis Clinical Research Network. Prednisone, azathioprine and N-acetylcysteine for pulmonary fi brosis. N Engl J Med 2012; 366: 1968–77.
127 Maher TM. Idiopathic pulmonary fibrosis: pathobiology of novel approaches to treatment. Clin Chest Med 2012; 33: 69–83.
128 Herazo-Maya JD, Noth I, Duncan SR, et al. Peripheral blood mononuclear cell gene expression profiles predict poor outcome in idiopathic pulmonary fibrosis. Sci Transl Med 2013; 5: 205ra136.
129 Yang IV, Coldren CD, Leach SM, et al. Expression of cilium-associated genes defi nes novel molecular subtypes of idiopathic pulmonary fi brosis. Thorax 2013; 68: 1114–21.
130 van’t Veer LJ, Dai H, van de Vijver MJ, et al. Gene expression profi ling predicts clinical outcome of breast cancer. Nature 2002; 415: 530–36.
131 Chen HY, Yu SL, Chen CH, et al. A fi ve-gene signature and clinical outcome in non-small-cell lung cancer. N Engl J Med 2007; 356: 11–20.
132 Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature 2009; 461: 747–53.
133 Yang IV, Schwartz DA. The next-generation of complex lung genetic studies. Am J Respir Crit Care Med 2012; 186: 1087–94.
134 Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004; 429: 457–63.
135 Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer 2004; 4: 143–53.
136 Yang IV. Epigenomics of idiopathic pulmonary fi brosis. Epigenomics 2012; 4: 195–203.
137 Dakhlallah D, Batte K, Wang Y, et al. Epigenetic regulation of miR-17~92 contributes to the pathogenesis of pulmonary fi brosis. Am J Respir Crit Care Med 2013; 187: 397–405.
138 Sanders YY, Ambalavanan N, Halloran B, et al. Altered DNA methylation profi le in idiopathic pulmonary fibrosis. Am J Respir Crit Car e Med 2012; 186: 525–35.
140. American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med. 2000; 161: 646–664
141. American Thoracic Society/European Respiratory Society. International multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2002; 165: 277–304
142. Raghu, G, Collard, HR, Egan, J et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011; 183: 788–824
143. Raghu, G, Weycker, D, Edelsberg, J, Bradford, WZ, and Oster, G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006; 174: 810–816
144. Hodgson, U, Laitinen, T, and Tukiainen, P. Nationwide prevalence of sporadic and familial idiopathic pulmonary fibrosis: evidence of founder effect among multiplex families in Finland. Thorax. 2002; 57: 338–342
145. Gribbin, J, Hubbard, RB, Le Jeune, I, Smith, CJ, West, J, and Tata, LJ. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax. 2006; 61: 980–985
146. van Moorsel, CH, van Oosterhout, MF, Barlo, NP et al. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a Dutch cohort. Am J Respir Crit Care Med. 2010; 182: 1419–1425
147. Alakhras, M, Decker, PA, Nadrous, HF, Collazo-Clavell, M, and Ryu, JH. Body mass index and mortality in patients with idiopathic pulmonary fibrosis. Chest. 2007; 131: 1448–1453
148. Gribbin, J, Hubbard, R, and Smith, C. Role of diabetes mellitus and gastro-oesophageal reflux in the aetiology of idiopathic pulmonary fibrosis. Respir Med. 2009; 103: 927–931
149. Lee, JS, Collard, HR, Raghu, G et al. Does chronic microaspiration cause idiopathic pulmonary fibrosis?. Am J Med. 2010; 123: 304–311
150. Nathan, SD, Basavaraj, A, Reichner, C et al. Prevalence and impact of coronary artery disease in idiopathic pulmonary fibrosis. Respir Med. 2010; 104: 1035–1041
151. Cottin, V, Le Pavec, J, Prevot, G et al. Pulmonary hypertension in patients with combined pulmonary fibrosis and emphysema syndrome. Eur Respir J. 2010; 35: 105–111
152. Ley, B, Collard, HR, and King, TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011; 183: 431–440
153. Song, JW, Hong, SB, Lim, CM, Koh, Y, and Kim, DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J. 2011; 37: 356–363
CrossRef | PubMed | Scopus (91)
154. Wootton, SC, Kim, DS, Kondoh, Y et al. Viral Infection in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2011; DOI: http://dx.doi.org/10.1164/rccm.201010-1752OC (published online Feb 25.)
155. Sisson, TH, Mendez, M, Choi, K et al. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am J Respir Crit Care Med. 2010; 181: 254–263
156. Seibold, MA, Wise, AL, Speer, MC et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011; 364: 1503–1512
157. Zhang, Y, Noth, I, Garcia, JGN, and Kaminski, N. A variant in the promoter of MUC5B and idiopathic pulmonary fibrosis. N Engl J Med. 2011; 364: 1576–1577
158. Pulkkinen, V, Bruce, S, Rintahaka, J et al. ELMOD2, a candidate gene for idiopathic pulmonary fibrosis, regulates antiviral responses. FASEB J. 2010; 24: 1167–1177
159. Walsh, DW, Godson, C, Brazil, DP, and Martin, F. Extracellular BMP-antagonist regulation in development and disease: tied up in knots. Trends Cell Biol. 2010; 20: 244–256
160. Marmai, C, Sutherland, RE, Kim, KK et al. Alveolar epithelial cells express mesenchymal proteins in patients with idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2011; DOI: http://dx.doi.org/10.1152/ajplung.00212.2010 (published online April 15.)
161. Maher, TM, Evans, IC, Bottoms, SE et al. Diminished prostaglandin E2 contributes to the apoptosis paradox in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010; 182: 73–82
162. Selman, M, Rojas, M, Mora, AL, and Pardo, A. Aging and interstitial lung diseases: unraveling an old forgotten player in the pathogenesis of lung fibrosis. Semin Respir Crit Care Med. 2010; 31: 607–617
163. Sharma, S, Kelly, TK, and Jones, PA. Epigenetics in cancer. Carcinogenesis. 2010; 31: 27–36
Lanceta, J, Prough, RA, Liang, R, and Wang, E. MicroRNA group disorganization in aging. Exp Gerontol. 2010; 45: 269–278
164. Pandit, KV, Corcoran, D, Yousef, H et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010; 182: 220–229
165. Liu, G, Friggeri, A, Yang, Y et al. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J Exp Med. 2010; 207: 1589–1597
166. Cushing, L, Kuang, PP, Qian, J et al. MIR-29 is a major regulator of genes associated with pulmonary fibrosis. Am J Respir Cell Mol Biol. 2010; DOI: http://dx.doi.org/10.1165/rcmb.2010-0323OC (published online Oct 22.)
167. King, TE Jr, Brown, KK, Raghu, G et al. BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011; DOI: http://dx.doi.org/10.1164/rccm.201011-1874OC (published online April 7.)
168. Daniels, CE, Lasky, JA, Limper, AH, Mieras, K, Gabor, E, and Schroeder, DR. Imatinib treatment for idiopathic pulmonary fibrosis: randomized placebo-controlled trial results. Am J Respir Crit Care Med. 2010; 181: 604–610
169. Taniguchi, H, Ebina, M, Kondoh, Y et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2010; 35: 821–829
170. Noble, PW, Albera, C, Bradford, WZ et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011; 377: 1760–1769
171. Crosby, LM and Waters, CM. Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol. 2010; 298: L715–L731
172. Wang, D, Morales, JE, Calame, DG, Alcorn, JL, and Wetsel, RA. Transplantation of human embryonic stem cell-derived alveolar epithelial type II cells abrogates acute lung injury in mice. Mol Ther. 2010; 18: 625–634
173. Petersen, TH, Calle, EA, Zhao, L et al. Tissue-engineered lungs for in vivo implantation. Science. 2010; 329: 538–541
174Zisman, DA, Schwarz, M, Anstrom, KJ, Collard, HR, Flaherty, KR, and Hunninghake, GW. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N Engl J Med. 2010; 363: 620–628

Debe leer
ResponderEliminarEste es un mensaje testimonial sobre mi padre que tiene 52 años y vivimos en Liverpool, Reino Unido. Lleva 3 años viviendo con fibrosis pulmonar. Desde su diagnóstico no ha sido fácil para nosotros ver a mi padre sufrir y no estaba contento con esta horrible enfermedad que tuvo que hacer cambios significativos en su vida. Sus síntomas son dificultad para respirar, sentirse cansado y toser, especialmente en la mañana. Lucha para escalar o caminar, se queda sin aliento fácilmente y muchos de sus efectos secundarios vinieron junto con sus medicamentos. Hemos intentado todo para encontrar una cura, pero el caso sigue empeorando con el tiempo.
Estuve en casa un buen domingo por la mañana cuando mi neumólogo me llamó debido a lo que hemos hablado acerca de todos estos años de estar abierto a cualquier tipo de sugerencia con respecto a la enfermedad de mi padre. Hace años que se ha confirmado la cura con la ayuda de un medicamento a base de hierbas llamado Malva-H, me pidió que me pusiera en contacto con el herborista a través de su blog; https: // curetoidiopathicpulmonaryfibrosis. blogspot. com o con su correo electrónico; dr.georgetom @ gmail. com, compré la medicina herbal y estoy feliz de decir que mi padre está siendo curado, contáctelo para IPF y COPD, el testimonio es para crear conciencia sobre el Dr. George y su medicina herbal Malva-H.