
Lípidos ectópicos, resistencia a la insulina y enfermedad cardiometabólica
Mecanismos íntimos moleculares: La causa más frecuente de depósito ectópico de lípidos en el músculo esquelético y en el hígado es el consumo de calorías superior al gasto calórico y la disminución de estos lípidos ectópicos se asocia con la neutralización de la resistencia a la insulina en estos órganos.
Autor(es): Shulman GI Fuente: N Engl J Med 2014; 371:1131-41.
Enlace: N Engl J Med 2014; 371:1131-41

Mecanismos íntimos moleculares: La causa más frecuente de depósito ectópico de lípidos en el músculo esquelético y en el hígado es el consumo de calorías superior al gasto calórico y la disminución de estos lípidos ectópicos se asocia con la neutralización de la resistencia a la insulina en estos órganos.
Autor(es): Shulman GI Fuente: N Engl J Med 2014; 371:1131-41.
Enlace: N Engl J Med 2014; 371:1131-41

En este artículo, el autor se centra sobre estudios recientes con espectroscopia por resonancia magnética (ERM) sobre la acumulación de lípidos ectópicos en la patogenia de la resistencia a la insulina en el músculo y el hígado.
Habría un posible vínculo entre la inflamación y la lipólisis inducida por macrófagos en la progresión de la resistencia a la insulina - inducida por lípidos ectópicos - a intolerancia a la glucosa y diabetes 2.
|
La diabetes tipo 2 afecta a gran número de personas en todo el mundo y es la causa principal de nefropatía terminal, de pérdida de un miembro no causada por traumatismo y de ceguera. Según las proyecciones, cabe esperar que la prevalencia mundial de diabetes tipo 2 aumente en más de un 75% durante las próximas dos décadas, en especial en Asia.
Aunque la disfunción de las células beta es responsable de la progresión de la normoglucemia a hiperglucemia, la resistencia a la insulina es anterior a esta disfunción y tiene una importante función en la patogenia de la diabetes tipo 2. Tras el consumo de hidratos de carbono, la glucosa se deposita principalmente en los músculos y en el hígado en forma de glucógeno y las alteraciones de la sensibilidad a la insulina en estos órganos producen hiperglucemia en ayunas y posprandial.
En este artículo, el autor se centra sobre estudios recientes con espectroscopia por resonancia magnética (ERM) sobre la acumulación de lípidos ectópicos en la patogenia de la resistencia a la insulina en el músculo y el hígado y dilucidaron la importancia de la resistencia a la insulina específica del músculo para favorecer el aumento de la lipogénesis hepática, el hígado graso no alcohólico y la dislipidemia aterógena. Propone después un posible vínculo entre la inflamación y la lipólisis inducida por macrófagos en la progresión de la resistencia a la insulina - inducida por lípidos ectópicos - a intolerancia a la glucosa y diabetes tipo 2.
Hipótesis del ciclo de los ácidos grasos en la resistencia a la insulina en el músculo
La asociación entre el depósito excesivo de lípidos en la obesidad y la resistencia a la insulina se conoce desde hace tiempo, y estudios ERM de protones (1H) mostraron una relación aún más estrecha entre el contenido intramiocelular de lípidos y la resistencia a la insulina en el músculo. No obstante, aún no hay acuerdo sobre el mecanismo molecular por el cual la grasa causa resistencia a la insulina. Hace más de un siglo, Randall y colaboradores propusieron la primera hipótesis, que posteriormente fue refutada por otros estudios.
La hipótesis actual es que la acumulación de un metabolito lipídico intracelular interviene en la resistencia a la insulina asociada con la obesidad y la diabetes tipo 2 al causar defectos en las señales de la insulina y disminuir la actividad de transporte de la glucosa estimulada por la insulina.
Mecanismos moleculares de la resistencia a la insulina en el músculo y en el hígado
La acción de la insulina en el músculo y el hígado exige la transmisión coordinada de las señales intracelulares que afectan sobre todo la fosforilación y la desfosforilación. En el músculo esquelético, la insulina fija y activa el receptor de insulina tirosinacinasa, con la ulterior fosforilación del sustrato 1 del receptor de insulina (IRS-1 por las siglas del inglés).
Una vez fosforilado, IRS-1 fija y activa la fosfatidilinositol 3-cinasa (PI3K), que a su vez, a través de intermediarios de señales, promueve la traslocación del transportador de glucosa tipo 4 (GLUT4) a la membrana celular, lo que produce la captación de glucosa en el músculo esquelético.
La fosforilación de la tirosina del IRS-1 estimulada por la insulina y la activación asociada de PI3K están alteradas en el músculo durante la infusión de lípidos en seres humanos y en roedores. Esto indica que la reducción inducida por lípidos en el transporte de la glucosa estimulado por la insulina se podría atribuir a un defecto proximal de las señales de insulina debido a una señal intracelular derivada de los ácidos grasos. Esta señal se identificó en estudios en roedores a los que se les administró una infusión de lípidos o se los alimentó con alto contenido de grasas. Los roedores tuvieron aumentos transitorios del contenido muscular de diacilglicerol (DAG) y activación sostenida de la forma teta de la proteína cinasa C (PKCθ), que activó la cascada de una serina– treonina y la inhibición de las señales de insulina.
En seres humanos se mostró que el contenido de DAG aumenta transitoriamente en el músculo esquelético tras la infusión de lípidos más heparina o de lípidos solos, El aumento del contenido de DAG en el músculo se asocia con aumentos en la actividad de PKCθ y la fosforilación de IRS-1 en Ser 1101.
Además, se observó aumento del contenido muscular de DAG, junto con aumento de la actividad de PKCθ y aumento de la fosforilación de la serina de IRS-1, en el músculo de personas obesas con resistencia a la insulina y personas con diabetes tipo 2.
La activación por DAG de una nPKC causa resistencia a la insulina tanto en el hígado como en el músculo. La esteatosis hepática y la resistencia hepática a la insulina aparecen en roedores tras sólo unos pocos días de alimentación abundante en grasas, sin cambios significativos en el contenido lipídico o la resistencia a la insulina en el músculo. En este modelo, la esteatosis hepática y la acumulación hepática de DAG se asociaron con defectos proximales de las señales de insulina con disminución de la fosforilación de la tirosina del IRS-1 e IRS-2 estimulada por la insulina y por el receptor de insulina, que finalmente interfiere con la activación de la síntesis de glucógeno y la supresión de la producción de glucosa en el hígado, inducidas por la insulina.
La alteración de la síntesis de glucógeno hepático estimulada por la insulina es similar a la de los pacientes con diabetes tipo 2. La asociación entre la activación de DAG–PKCε en el hígado y la resistencia hepática a la insulina se mostró en numerosos modelos de roedores transgénicos con hígado graso no alcohólico. Más importante, el aumento del contenido hepático de DAG y el aumento de la actividad de PKCε son los factores pronósticos más fuertes de resistencia hepática a la insulina en personas obesas con hígado graso no alcohólico.
Disociación entre la obesidad y la resistencia a la insulina en el músculo y en el hígado
La causa más frecuente de depósito ectópico de lípidos en el músculo esquelético y el hígado es el consumo de calorías superior al gasto calórico. Al contrario de la obesidad, las lipodistrofias ofrecen la oportunidad de evaluar la función del depósito de lípidos ectópicos sin la contribución de ninguna expansión de masa de tejido adiposo periférico o visceral. La falta de grasa subcutánea lleva a la hipertrigliceridemia, el depósito ectópico de grasa (incluida una considerable esteatosis hepática) y una profunda resistencia a la insulina en el músculo y el hígado.
En ratones lipoatróficos A-ZIP/F-1, que no tienen adipocitos, se acumula grasa en el hígado y el músculo esquelético, tejidos en los que se produce una profunda resistencia a la insulina. Si se les trasplanta grasa por vía subcutánea, se normaliza el contenido lipídico ectópico en el músculo y el hígado, así como las señales y la acción de la insulina en estos órganos.
Otra evidencia que apoya la importancia de la acumulación de lípidos ectópicos en la patogenia de la resistencia a la insulina en el músculo y el hígado surge de estudios en ratones transgénicos. Ratones transgénicos con sobrexpresión de la lipoproteína lipasa en el hígado tienen acumulación de grasa específica para el hígado y resistencia a la insulina específica para el hígado.
Asimismo, ratones transgénicos con sobrexpresión de la lipoproteína lipasa en el músculo esquelético tienen acumulación de grasa específica para el músculo y resistencia a la insulina específica para el músculo. En conjunto, estos estudios muestran que la acumulación ectópica de lípidos intracelulares conduce a la resistencia a la insulina en el músculo y el hígado, incluso en ausencia de adiposidad periférica y visceral y que los DAG son los metabolitos derivados de los lípidos responsables de desencadenar la resistencia a la insulina mediante la activación de PKCε en el hígado y PKCθ en el músculo.
En unas pocas excepciones, la acumulación de lípidos ectópicos en el músculo y el hígado se disocia de la resistencia a la insulina. Una de estas excepciones es el síndrome de Chanarin–Dorfman, debida a la deficiencia de la proteína llamada identificación de genes comparativa 58 (CGI-58). Estudios mostraron que la compartimentación de los DAG dentro de las gotitas lipídicas es la probable explicación de la disociación entre la acumulación de lípidos ectópicos y la resistencia a la insulina en este síndrome. Aún no se sabe si la explicación es la misma para la disociación entre el aumento de la acumulación de líquidos ectópicos y la resistencia a la insulina en otros casos, como la hipobetalipoproteinemia familiar y en el músculo de los atletas de resistencia.
Importancia de la disfunción mitocondrial en la acumulación de lípidos ectópicos
El contenido lipídico de las células musculares refleja un equilibrio entre la captación de ácidos grasos por las células y la oxidación de las grasas mitocondriales. La disfunción mitocondrial adquirida es un importante factor predisponente para la acumulación ectópica de lípidos y la resistencia a la insulina en los ancianos. Se halló que en personas ancianas, delgadas, la captación de glucosa estimulada por la insulina era mucho menor que la de personas jóvenes emparejadas para la masa magra y la masa grasa.
En ancianos, la resistencia a la insulina en el músculo se asoció con aumento de la acumulación lipídica en las células musculares y reducción de aproximadamente el 40% en la actividad oxidativa y de fosforilación mitocondrial, evaluada mediante ERM C y P in vivo, en relación con la actividad oxidativa y de fosforilación en controles jóvenes. Estos datos avalan la hipótesis de que las reducciones de la función mitocondrial asociadas con la edad, posiblemente debidas a daño acumulado por las especies reactivas del oxígeno (ERO), predisponen a los ancianos a la acumulación ectópica de lípidos y la resistencia a la insulina en el músculo.
Estudios en ratones transgénicos con sobrexpresión de catalasa humana dirigida hacia las mitocondrias apoyan esta hipótesis.
En conjunto, estos datos muestran que las reducciones adquiridas de la función mitocondrial asociadas con la edad promueven la acumulación ectópica de lípidos en el músculo esquelético y la resistencia a la insulina en el músculo. También sugieren que conservar la función mitocondrial mediante la reducción del daño oxidativo mitocondrial puede ser un objetivo terapéutico para prevenir la disminución de la función mitocondrial en el músculo asociada con la edad, la resistencia a la insulina en el músculo y la diabetes tipo 2 en los ancianos.
Disminuciones de aproximadamente el 40% en la actividad oxidativa y de fosforilación mitocondrial en el músculo se observaron en jóvenes sanos, delgados, con resistencia a la insulina, cuyos padres padecen diabetes tipo 2. La disminución del flujo en el ciclo del ácido tricarboxílico y de la síntesis del ATP en el músculo fue paralela a la disminución de aproximadamente el 40% del contenido mitocondrial.
Es así probable que la reducción del contenido mitocondrial, debida a la disminución de la biogénesis mitocondrial, sea responsable de la reducción de la actividad oxidativa y de fosforilación de las mitocondrias y puede ser una alteración adquirida. No obstante, dada la función clave de la actividad mitocondrial en la regulación del metabolismo del as grasas en las células musculares, estos datos sugieren que la función mitocondrial reducida puede ser un importante factor predisponente que promueve la acumulación de DAG en las células musculares y la resistencia a la insulina en el músculo en personas con resistencia a la insulina cuyos padres padecen diabetes tipo 2.
Alteraciones que favorecen la acumulación de lípidos ectópicos en el hígado
Aunque el hígado graso no alcohólico casi siempre se asocia con obesidad, hay excepciones importantes a esta regla, donde se observan hígado graso no alcohólico y resistencia hepática a la insulina en personas delgadas.
Polimorfismos en la respuesta a la insulina del gen que codifica la apolipoproteína C3 (APOC3) predisponen a estas personas al hígado graso no alcohólico y la resistencia hepática a la insulina.
Estos polimorfismos generan un aumento del 30% de la apolipoproteína C3 plasmática. Este aumento inhibe la actividad de la lipoproteína lipasa, limitando la depuración periférica de quilomicrones y causando hipertrigliceridemia posprandial. Como resultado, los portadores de las variantes de APOC3 tienen aumento de la captación hepática de lípidos de los restos de los quilomicrones, que los predisponen al hígado graso no alcohólico y a la resistencia hepática a la insulina.
La evidencia genética en apoyo de la importancia de las alteraciones de la apolipoproteína en la regulación de la síntesis hepática de triglicéridos proviene de estudios en ratones transgénicos con sobreexpresión de la apolipoproteína C3 en el hígado.
Estos estudios sugieren que las interacciones entre genes y medioambiente pueden predisponer a personas delgadas al hígado graso no alcohólico, la resistencia hepática a la insulina y la diabetes tipo 2 y estas interacciones pueden también afectar muchas posibles variantes de las apolipoproteínas plasmáticas que involucran la actividad de la lipoproteína lipasa. La interacción gen-ambiente de la APOC3 se observó sólo en hombres. Es probable que esto refleje un efecto protector del estradiol sobre la capacidad de la apolipoproteína C3 para inhibir la actividad de la lipoproteína lipasa y favorecer el depósito de grasa ectópica en mujeres premenopáusicas.
Además, la interacción gen APOC3- ambiente no se observa en personas obesas; éstas tienen esteatosis hepática, que enmascara el efecto relativamente sutil de las variantes de APOC3 para predisponer a las personas al hígado graso y la resistencia hepática a la insulina.
Los hispanoamericanos son otro gran grupo étnico en riesgo de hígado graso no alcohólico, resistencia a la insulina y diabetes tipo 2. Se identificó una mutación de aminoácido (I148 M in PNPLA3) que es más frecuente en hispanoamericanos que en otros grupos étnicos y se asocia fuertemente con el hígado graso.
Por último, los genes que regulan la lipogénesis (e.g., AGPAT2 y PPARG), y llevan a la lipodistrofia, y las alteraciones de los genes que regulan la lipólisis (e.g., los genes que codifican la perilipina [PLIN1]) también generan acumulación de lípidos ectópicos y resistencia a la insulina.
Neutralización de la resistencia a la insulina y la diabetes mediante la reducción de la grasa ectópica
Otra evidencia de que la acumulación de lípidos ectópicos en el músculo y el hígado tiene una función causal en la patogenia de la resistencia a la insulina y la diabetes tipo 2 proviene de estudios que muestran que la disminución del contenido de lípidos ectópicos se asocia con la neutralización de la resistencia a la insulina en estos órganos. Un estudio indicó que el restablecimiento de la leptina plasmática a niveles fisiológicos en pacientes con diabetes y lipodistrofia normalizó la glucemia en ayunas y las concentraciones plasmáticas de lípidos.
Estas mejorías del metabolismo de la glucosa estimulado por la insulina, que se pueden atribuir a la neutralización de la resistencia a la insulina en el músculo y el hígado, se asociaron con grandes disminuciones del contenido hepático de triglicéridos y del contenido de grasa en los miocitos.
Asimismo, el descenso de aproximadamente el 10% del peso corporal con una dieta hipocalórica disminuyó notablemente las concentraciones de triglicéridos hepáticos y normalizó la sensibilidad hepática a la insulina, la producción hepática de glucosa y la glucemia en ayunas en pacientes con diabetes tipo 2.
Se observaron disminuciones de la grasa de los miocitos y neutralización de la resistencia a la insulina en el músculo tras el descenso de aproximadamente el 10% del peso en personas jóvenes y delgadas con resistencia a la insulina cuyos padres sufrían diabetes tipo 2.
Las tiazolidinedionas también disminuyen la esteatosis hepática y mejoran la sensibilidad a la insulina en el músculo y el hígado, al aumentar la sensibilidad a la insulina de los adipositos y desplazar los lípidos ectópicos del músculo y el hígado al tejido adiposo subcutáneo.
Resistencia a la insulina en el músculo esquelético, dislipidemia e hígado graso no alcohólico
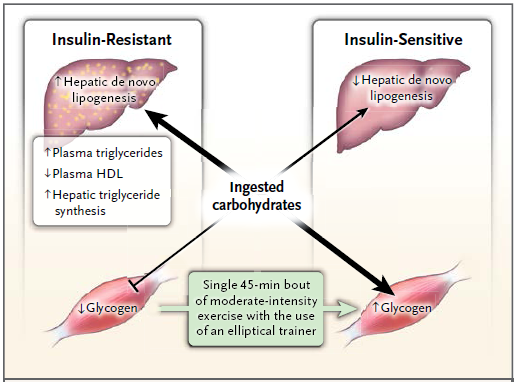
El aumento de las grasas en los miocitos y la resistencia a la insulina en el músculo esquelético son defectos iniciales observados en la patogenia de la diabetes tipo 2.
En personas jóvenes sanas, la resistencia selectiva a la insulina en el músculo favorece la dislipidemia aterógena al cambiar el patrón de los hidratos de carbono consumidos, de la síntesis de glucógeno en el musculo esquelético a la lipogénesis hepática de novo, produciendo aumento de los triglicéridos plasmáticos y disminución de las lipoproteínas de alta densidad.
Además, este patrón anormal de almacenamiento energético fue suprimido por completo tras un único episodio de ejercicio de intensidad moderada con la elíptica, que favoreció la síntesis de glucógeno muscular tras el consumo de hidratos de carbono, mediante el aumento de la actividad de transporte de la glucosa. Estos datos muestran que la resistencia a la insulina en el músculo es un objetivo inicial para el tratamiento y la prevención de la dislipidemia aterógena y del hígado graso no alcohólico en personas jóvenes con resistencia a la insulina, que son propensas al síndrome metabólico y la diabetes tipo 2.
 EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG |
Lipólisis inducida por los macrófagos, inflamación e hiperglucemia en ayunas
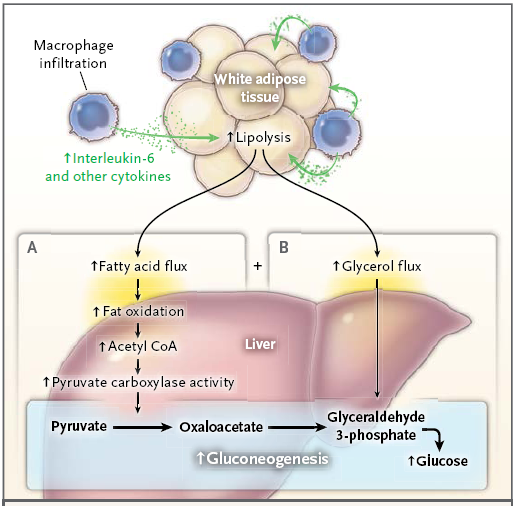
Aunque la resistencia a la insulina inducida por lípidos aparece precozmente en la patogenia de la diabetes tipo 2, es esencial identificar los factores que promueven la progresión de la resistencia a la insulina asociada con la acumulación de lípidos ectópicos a la intolerancia a la glucosa y la hiperglucemia en ayunas.
El punto de vista tradicional atribuye la alteración de la función las células pancreáticas beta y alfa, junto con la inflamación, a esta transición, en la que defectos de las células beta y alfa llevan al aumento de la transcripción de genes gluconeogénicos hepáticos y la inflamación inhibe la acción de la insulina a través de la liberación de citocinas y adipocitocinas.
El aumento de las citocinas a su vez causa inhibición de las señales de insulina y aumento de la transcripción de la proteína gluconeogénica hepática a través de la activación del factor nuclear kβ, la cinasa Jun N-terminal y las vías biosíntéticas de la ceramida.
Otra hipótesis que vincula la inflamación con la progresión a la hiperglucemia en ayunas es el posible efecto de la lipólisis inducida por macrófagos sobre la regulación de la gluconeogénesis hepática. Estudios en ratones avalan esta hipótesis. Aunque especulativa, esta hipótesis propone que la lipólisis inducida por macrófagos, al contrario de las alteraciones en las citocinas circulantes y la transcripción de la proteína gluconeogénica hepática, es el principal culpable de la transición de la resistencia a la insulina a la intolerancia a la glucosa y la diabetes tipo 2. Esta hipótesis también coincide con un estudio que mostró la ausencia de relación entre la expresión de la proteína gluconeogénica
hepática y la hiperglucemia en ayuna en personas obesas.
 EMERGENCY & CRITICAL CARE WITH DR. RAFAEL PEREZ GARCIA® HEALTH BLOG |
Posibles tratamientos para la acumulación ectópica de lípidos y la resistencia a la insulina
La resistencia a la insulina inducida por lípidos ectópicos representa un exceso de energía intracelular en forma de DAG, que genera la activación de PKCθ en el músculo y de PKCε en el hígado y la ulterior inhibición de las señales de insulina en estos tejidos. Esta hipótesis puede explicar la resistencia a la insulina asociada con la obesidad, el envejecimiento, la lipodistrofia, la prediabetes y la diabetes tipo 2 y la neutralización de la resistencia a la insulina y de la diabetes tras el descenso de peso y el tratamiento con tiazolidinediona.
Teleológicamente, la resistencia a la insulina en el músculo y el hígado inducida por DAG y nPKCs puede representar un mecanismo celular autónomo para detener el depósito de energía en el hígado y el músculo ante el exceso de lípidos intracelulares y encauzar ese exceso de energía al tejido adiposo para su almacenamiento.
Aunque la reducción del contenido de lípidos ectópicos y de la resistencia a la insulina mediante intervenciones para el descenso de peso (idealmente junto con ejercicio) es el tratamiento preferido para estos trastornos, la recidiva tras el adelgazamiento es muy frecuente. La cirugía bariátrica es más exitosa para lograr el descenso de peso a largo plazo, pero es un procedimiento invasivo, caro y con ciertos riesgos. Por consiguiente, se necesita un fármaco que disminuya la grasa ectópica del hígado y la resistencia a la insulina.
El factor de crecimiento fibroblástico 21 es eficaz para reducir la actividad hepática de DAG–PKCε, así como la resistencia hepática a la insulina en animales y actualmente se lo está investigando en ensayos clínicos.
Otro enfoque para disminuir el contenido de lípidos ectópicos es la aplicación de protonoforo mitocondrial dirigido al hígado para favorecer aumentos sutiles del desacoplamiento mitocondrial hepático.
Este enfoque neutraliza la hipertrigliceridemia, la esteatosis hepática, la resistencia a la insulina y la hiperglucemia en modelos murinos de hígado graso no alcohólico y diabetes tipo 2. Además de disminuir los triglicéridos y el DAG hepático, la actividad de PKCε y la resistencia hepática a la insulina, este enfoque reduce el contenido hepático de acetil CoA, disminuye la gluconeogénesis hepática y la hiperglucemia en ayunas y posprandial. Además, al disminuir la oxidación de la grasa hepática en un 60%, disminuye la producción hepática de lipoproteína de muy baja densidad y produce así menor pasaje de triglicéridos al músculo y protege contra la resistencia a la insulina en el músculo inducida por lípidos.
En resumen, estos estudios muestran la importancia de la acumulación ectópica de lípido en la patogenia de la resistencia a la insulina en el músculo y el hígado. Este modelo también explica la mejor acción de la insulina con el ejercicio, el adelgazamiento y las tiazolidinedionas. Asimismo, aumentar el gasto hepático de energía al favorecer el desacoplamiento mitocondrial podría ser un enfoque novedoso para tratar la epidemia de hígado graso no alcohólico, el síndrome metabólico y la diabetes tipo 2.
►Artículo ➔ Noticia ➲ Tema básico ➜ Editorial
1. International Diabetes Federation. IDF diabetes atlas. 6th ed. Brussels: International Diabetes Federation, 2014 (http://www.idf.org/diabetesatlas).
2. Porte D Jr, Kahn SE. Beta-cell dysfunction and failure in type 2 diabetes: potential mechanisms. Diabetes 2001;50: Suppl 1:S160-S163.
3. Rothman DL, Magnusson I, Cline G, et al. Decreased muscle glucose transport/ phosphorylation is an early defect in the pathogenesis of non-insulin-dependent diabetes mellitus. Proc Natl Acad Sci U S A 1995; 92:983-7.
4. Shulman GI, Rothman DL, Jue T, Stein P, DeFronzo RA, Shulman RG. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non–insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N Engl J Med 1990; 322:223-8.
5. Taylor R, Magnusson I, Rothman DL, et al. Direct assessment of liver glycogen storage by 13C nuclear magnetic resonance spectroscopy and regulation of glucose homeostasis after a mixed meal in normal subjects. J Clin Invest 1996; 97: 126-32.
6. Krssak M, Falk Petersen K, Dresner A, et al. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia 1999; 42:113-6. [Errata, Diabetologia 1999;42:386, 1269.]
7. Perseghin G, Scifo P, De Cobelli F, et al. Intramyocellular triglyceride content is a determinant of in vivo insulin resistance in humans: a 1H-13C nuclear magnetic resonance spectroscopy assessment in offspring of type 2 diabetic parents. Diabetes 1999; 48:1600-6.
8. Stefan N, Kantartzis K, Machann J, et al. Identification and characterization of metabolically benign obesity in humans. Arch Intern Med 2008; 168:1609-16.
9. Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle: its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963;1: 785-9.
10. Roden M, Price TB, Perseghin G, et al. Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest 1996; 97:2859-65.
11. Dresner A, Laurent D, Marcucci M, et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest 1999; 103:253-9.
12. Petersen KF, Hendler R, Price T, et al. 13C/31P NMR studies on the mechanism of insulin resistance in obesity. Diabetes 1998;47: 381-6.
13. Cline GW, Petersen KF, Krssak M, et al. Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. N Engl J Med 1999; 341:240-6.
14. Perseghin G, Price TB, Petersen KF, et al. Increased glucose transport–phosphorylation and muscle glycogen synthesis after exercise training in insulin-resistant subjects. N Engl J Med 1996; 335: 1357-62.
15. Griffin ME, Marcucci MJ, Cline GW, et al. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes 1999; 48:1270-4.
16. Yu C, Chen Y, Cline GW, et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem 2002; 277:50230-6.
17. Kim JK, Fillmore JJ, Sunshine MJ, et al. PKC-theta knockout mice are protected from fat-induced insulin resistance. J Clin Invest 2004; 114:823-7.
18. Morino K, Neschen S, Bilz S, et al. Muscle-specific IRS-1 Ser→Ala transgenic mice are protected from fat-induced insulin resistance in skeletal muscle. Diabetes 2008; 57:2644-51.
19. Li Y, Soos TJ, Li X, et al. Protein kinase C Theta inhibits insulin signaling by phosphorylating IRS1 at Ser(1101). J Biol Chem 2004; 279:45304-7.
20. Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes 2002; 51: 2005-11.
21. Szendroedi J, Yoshimura T, Phielix E, et al. Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. Proc Natl Acad Sci U S A 2014; 111:9597-602.
22. Morino K, Petersen KF, Dufour S, et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J Clin Invest 2005;115: 3587-93.
23. Corbould A, Kim YB, Youngren JF, et al. Insulin resistance in the skeletal muscle of women with PCOS involves intrinsic and acquired defects in insulin signaling. Am J Physiol Endocrinol Metab 2005;288: E1047-E1054.
24. Itani SI, Pories WJ, Macdonald KG, Dohm GL. Increased protein kinase C theta in skeletal muscle of diabetic patients. Metabolism 2001; 50:553-7.
25. Samuel VT, Liu ZX, Qu X, et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem 2004; 279:32345-53.
26. Krssak M, Brehm A, Bernroider E, et al. Alterations in postprandial hepatic glycogen metabolism in type 2 diabetes. Diabetes 2004; 53:3048-56.
27. Magnusson I, Rothman DL, Katz LD, Shulman RG, Shulman GI. Increased rate of gluconeogenesis in type II diabetes mellitus: a 13C nuclear magnetic resonance study. J Clin Invest 1992; 90:1323-7.
28. Choi CS, Savage DB, Abu-Elheiga L, et al. Continuous fat oxidation in acetyl-CoA carboxylase 2 knockout mice increases total energy expenditure, reduces fat mass, and improves insulin sensitivity. Proc Natl Acad Sci U S A 2007; 104:16480-5.
29. Matsuzaka T, Shimano H, Yahagi N, et al. Crucial role of a long-chain fatty acid elongase, Elovl6, in obesity-induced insulin resistance. Nat Med 2007; 13:1193-202.
30. Savage DB, Choi CS, Samuel VT, et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J Clin Invest 2006; 116:817-24.
31. Varela GM, Antwi DA, Dhir R, et al. Inhibition of ADRP prevents diet-induced insulin resistance. Am J Physiol Gastrointest Liver Physiol 2008; 295:G621-G628.
32. Zhang D, Liu Z-X, Choi CS, et al. Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc Natl Acad Sci U S A 2007; 104:17075-80.
33. Kumashiro N, Erion DM, Zhang D, et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A 2011; 108:16381-5.
34. Magkos F, Su X, Bradley D, et al. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology 2012; 142(7):1444.e2-1446.e2.
35. Raddatz K, Turner N, Frangioudakis G, et al. Time-dependent effects of Prkce deletion on glucose homeostasis and hepatic lipid metabolism on dietary lipid oversupply in mice. Diabetologia 2011;54: 1447-56.
36. Petersen KF, Oral EA, Dufour S, et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest 2002; 109:1345-50.
37. Lee HY, Choi CS, Birkenfeld AL, et al.Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab 2010; 12:668-74.
38. Petersen KF, Befroy D, Dufour S, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 2003; 300:1140-2.
39. Morino K, Petersen KF, Sono S, et al. Regulation of mitochondrial biogenesis by lipoprotein lipase in muscle of insulin resistant offspring of parents with type 2 diabetes. Diabetes 2012; 61:877-87.
40. Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med 2004; 350:664-71.
41. Kim JK, Gavrilova O, Chen Y, Reitman ML, Shulman GI. Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J Biol Chem 2000; 275:8456-60.
42. Kim JK, Fillmore JJ, Chen Y, et al. Tissue-specific overexpression of lipoprotein lipase causes tissue-specific insulin resistance. Proc Natl Acad Sci U S A 2001;98:7522-7.
43. Ferreira LD, Pulawa LK, Jensen DR, Eckel RH. Overexpressing human lipoprotein lipase in mouse skeletal muscle is associated with insulin resistance. Diabetes 2001; 50:1064-8. [Erratum, Diabetes 2001; 50:1512.]
44. Amaro A, Fabbrini E, Kars M, et al. Dissociation between intrahepatic triglyceride content and insulin resistance in familial hypobetalipoproteinemia. Gastroenterology 2010; 139:149-53.
45. Cantley JL, Yoshimura T, Camporez JP, et al. CGI-58 knockdown sequesters diacylglycerols in lipid droplets/ER-preventing diacylglycerol-mediated hepatic insulin resistance. Proc Natl Acad Sci U S A 2013; 110:1869-74.
46. Farese RV Jr, Zechner R, Newgard CB, Walther TC. The problem of establishing relationships between hepatic steatosis and hepatic insulin resistance. Cell Metab 2012; 15:570-3.
47. Amati F, Dubé JJ, Alvarez-Carnero E, et al. Skeletal muscle triglycerides, diacylglycerols, and ceramides in insulin resistance: another paradox in endurancetrained athletes? Diabetes 2011; 60: 2588-97.
48. Boumezbeur F, Mason GF, de Graaf RA, et al. Altered brain mitochondrial metabolism in healthy aging as assessed by in vivo magnetic resonance spectroscopy. J Cereb Blood Flow Metab 2010; 30: 211-21.
49. Zong H, Ren JM, Young LH, et al. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A 2002; 99:15983-7.
50. Reznick RM, Zong H, Li J, et al. Aging associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab 2007; 5:151-6.
51. Befroy DE, Petersen KF, Dufour S, et al. Impaired mitochondrial substrate oxidation in muscle of insulin-resistant offspring of type 2 diabetic patients. Diabetes 2007; 56:1376-81.
52. Sleigh A, Raymond-Barker P, Thackray K, et al. Mitochondrial dysfunction in patients with primary congenital insulin resistance. J Clin Invest 2011; 121:2457-61.
53. Choi CS, Fillmore JJ, Kim JK, et al. Overexpression of uncoupling protein 3 in skeletal muscle protects against fatinduced insulin resistance. J Clin Invest 2007; 117:1995-2003.
54. Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002; 51:2944-50.
55. Angulo P. Nonalcoholic fatty liver disease. N Engl J Med 2002; 346:1221-31.
56. Petersen KF, Dufour S, Befroy D, Lehrke M, Hendler RE, Shulman GI. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 2005; 54:603-8.
57. Petersen KF, Dufour S, Feng J, et al. Increased prevalence of insulin resistance and nonalcoholic fatty liver disease in Asian-Indian men. Proc Natl Acad Sci U S A 2006; 103:18273-7.
58. Petersen KF, Dufour S, Hariri A, et al. Apolipoprotein C3 gene variants in nonalcoholic fatty liver disease. N Engl J Med 2010; 362:1082-9.
59. Peter A, Kantartzis K, Machicao F, et al. Visceral obesity modulates the impact of apolipoprotein C3 gene variants on liver fat content. Int J Obes (Lond) 2012; 36:774-82.
60. Lee HY, Birkenfeld AL, Jornayvaz FR, et al. Apolipoprotein CIII overexpressing mice are predisposed to diet-induced hepatic steatosis and hepatic insulin resistance. Hepatology 2011; 54:1650-60.
61. Camporez JP, Jornayvaz FR, Lee HY, et al. Cellular mechanism by which estradiol protects female ovariectomized mice from high-fat diet-induced hepatic and muscle insulin resistance. Endocrinology 2013; 154:1021-8.
62. Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008; 40:1461-5.
63. Cortés VA, Curtis DE, Sukumaran S, et al. Molecular mechanisms of hepatic steatosis and insulin resistance in the AGPAT2-deficient mouse model of congenital generalized lipodystrophy. Cell Metab 2009; 9:165-76.
64. Gandotra S, Le Dour C, Bottomley W, et al. Perilipin deficiency and autosomal dominant partial lipodystrophy. N Engl J Med 2011; 364:740-8.
65. Lim EL, Hollingsworth KG, Aribisala BS, Chen MJ, Mathers JC, Taylor R. Reversal of type 2 diabetes: normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia 2011; 54:2506-14.
66. Petersen KF, Dufour S, Morino K, Yoo PS, Cline GW, Shulman GI. Reversal of muscle insulin resistance by weight reduction in young, lean, insulin-resistant offspring of parents with type 2 diabetes. Proc Natl Acad Sci U S A 2012; 109:8236-40.
67. Mayerson AB, Hundal RS, Dufour S, et al. The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes. Diabetes 2002; 51:797-802.
68. Belfort R, Harrison SA, Brown K, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med 2006; 355:2297-307.
69. Perseghin G, Ghosh S, Gerow K, Shulman GI. Metabolic defects in lean nondiabetic offspring of NIDDM parents: a cross-sectional study. Diabetes 1997; 46:1001-9.
70. Petersen KF, Dufour S, Savage DB, et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci U S A 2007; 104:12587-94.
71. Rab.l R, Petersen KF, Dufour S, Flannery C, Shulman GI. Reversal of muscle insulin resistance with exercise reduces postprandial hepatic de novo lipogenesis in insulin resistant individuals. Proc Natl Acad Sci U S A 2011; 108:13705-9.
72. Perry RJ, Zhang X-M, Zhang D, et al. Leptin reverses diabetes by suppression of the hypothalamic–pituitary–adrenal axis. Nat Med 2014; 20:759-63.
73. Samuel VT, Beddow SA, Iwasaki T, et al. Fasting hyperglycemia is not associated with increased expression of PEPCK or G6Pc in patients with type 2 diabetes. Proc Natl Acad Sci U S A 2009; 106:12121-6.
74. Camporez JP, Jornayvaz FR, Petersen MC, et al. Cellular mechanisms by which FGF21 improves insulin sensitivity in male mice. Endocrinology 2013; 154:3099-109.
75. Perry RJ, Kim T, Zhang XM, et al. Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver targeted mitochondrial uncoupler. Cell Metab 2013; 18:740-8.

No hay comentarios:
Publicar un comentario